2.1. Lewis Acid—Lewis Base Adducts
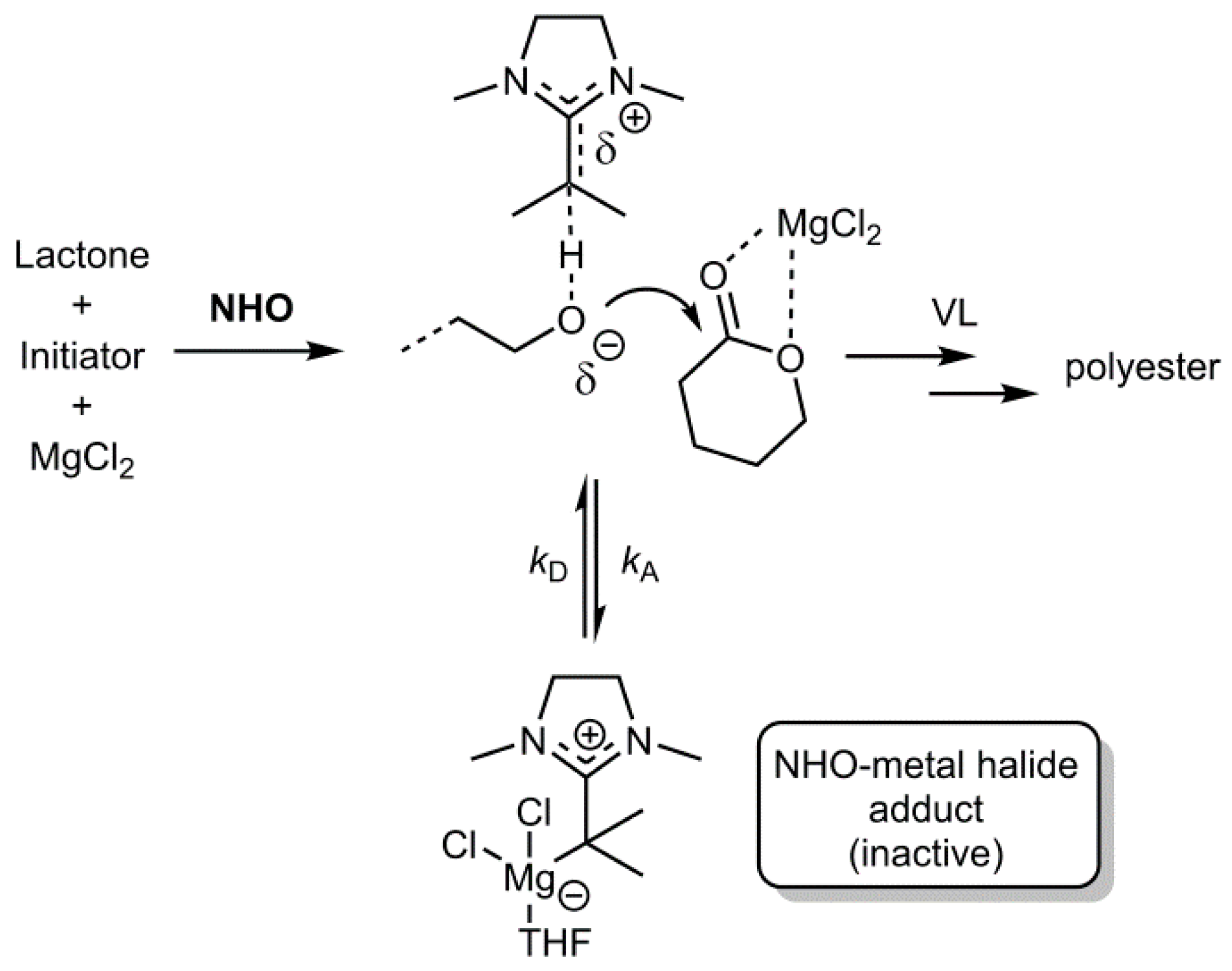
As a first step, NHO-adduct formation was investigated. Since the currently proposed polymerization mechanisms [
11,
12] are based on the assumption of a free NHO acting in cooperation with a free, i.e., non-complexed, Lewis acid, a sufficient lability of potential NHO-metal complexes in a given LP polymerization system must be ensured. The formation of too stable adducts would quench any polymerization activity; on the other hand, the choice of a too weakly coordinating metal ion will also impair its ability to activate lactones for ring-opening—an issue that has been identified as central in several polymerization systems employing
N-heterocyclic carbenes (NHCs) or NHOs [
4,
10,
11,
12,
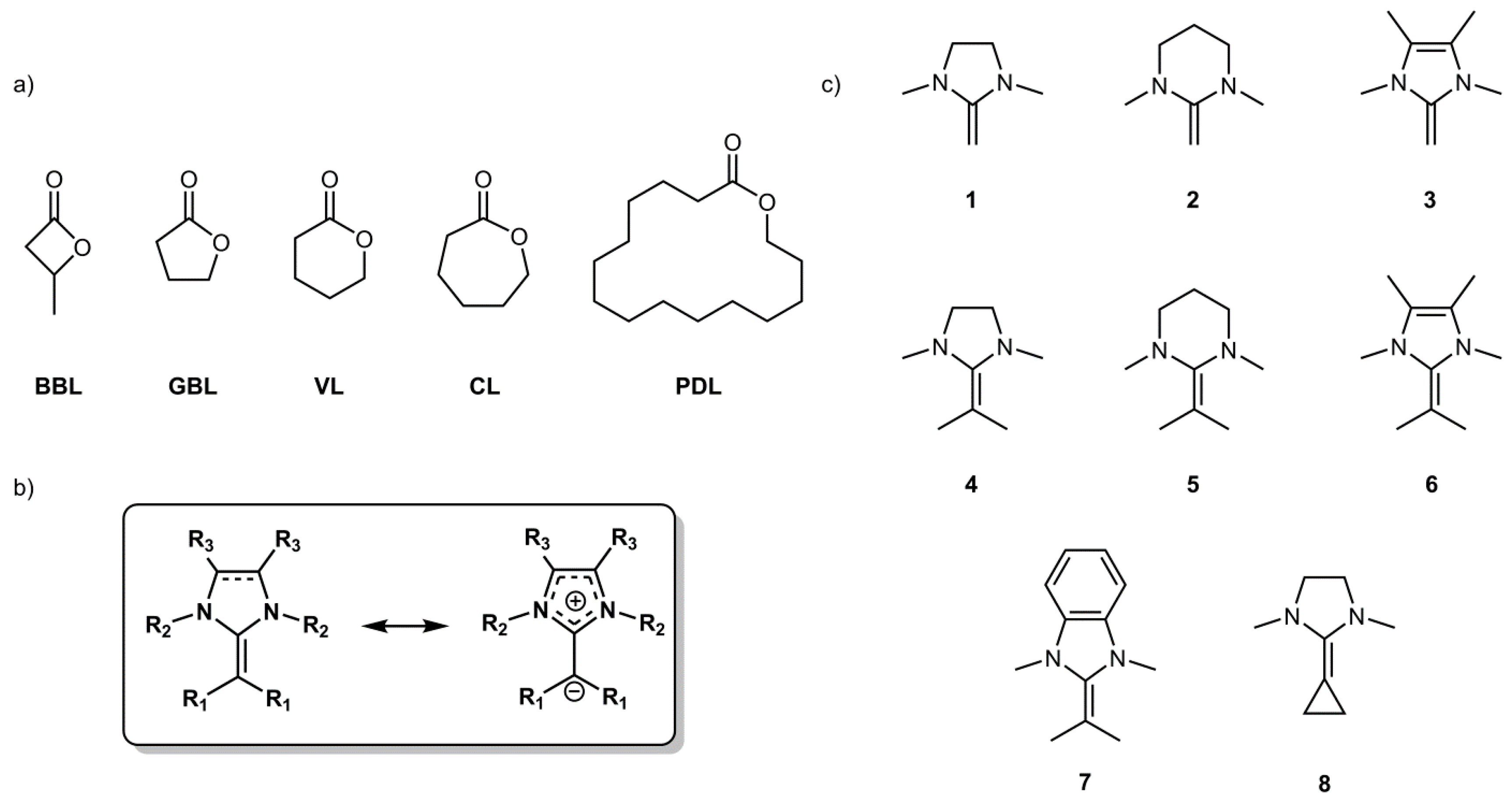
32]. Thus, in order to achieve a better understanding of this required balance, the affinity of eight structurally different NHOs (
1–
8,
Figure 1) to bind to each of the Lewis acids MgCl
2, ZnCl
2 and LiCl was investigated. Adduct geometries were optimized and the Gibbs Free Energy of the reaction (Δ
GR) as shown in
Scheme 2 was compared. Furthermore, the metal–C bond distances were investigated to obtain additional insight into the strength of the interaction. It should be noted that for the considered reaction, the NHO substitutes a tetrahydrofurane (THF) ligand; the generated complex is neutral, yet a charge separation takes place, locating the positive charge on the NHO ligand. A tetrahedral complex geometry was employed and it was assumed that the metal–Cl bond will resist substitution. THF is a typical solvent for lactone polymerization and the existence of such tetrahedral structures has been demonstrated for closely related NHC complexes such as ZnCl
2(SIMes)(THF) [
4]. The results for MgCl
2, ZnCl
2, and LiCl are shown in
Table 1,
Table 2 and
Table 3, respectively.
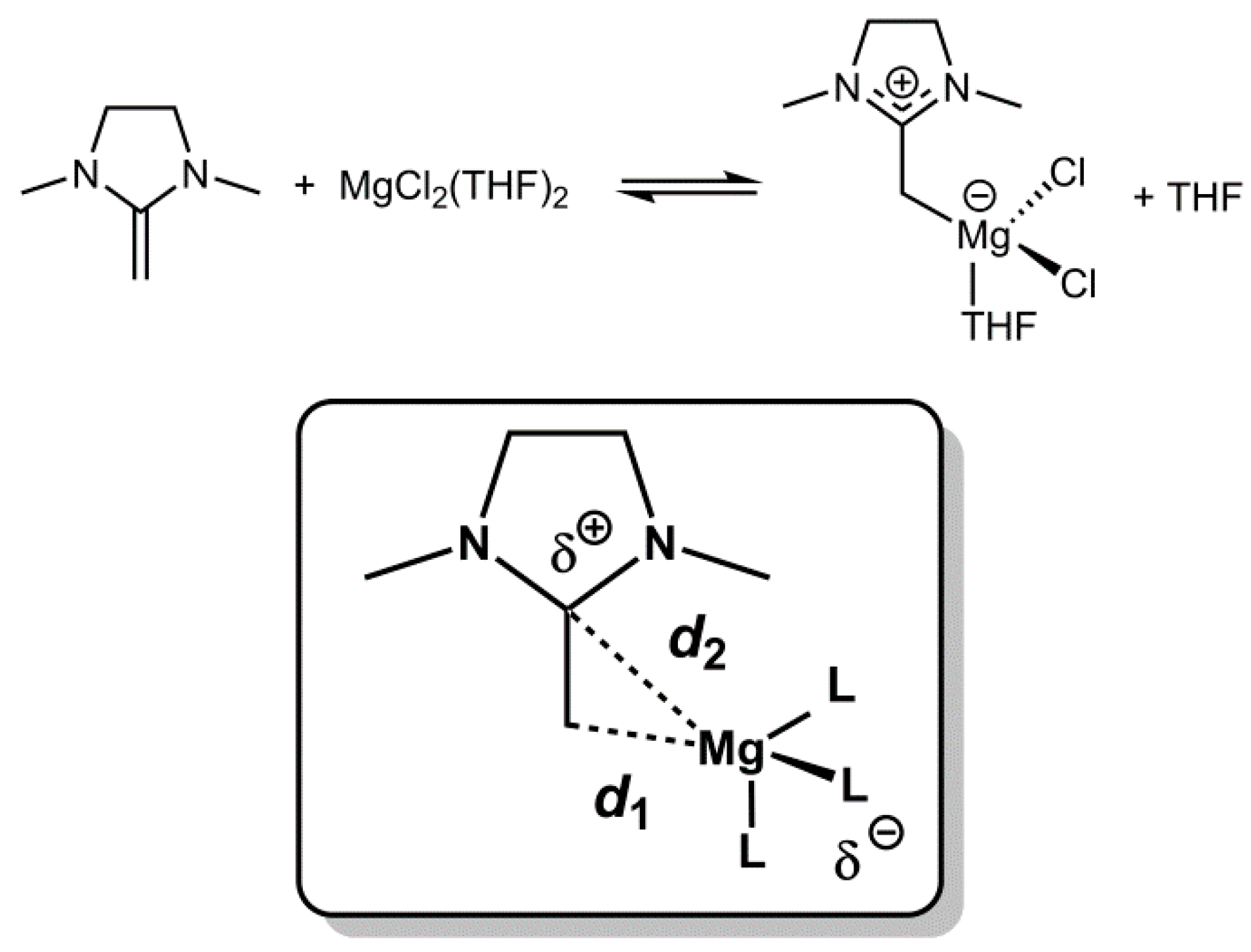
From these data, a number of conclusions can be drawn. As a consequence of the strong polarization of the olefinic double bond, the NHOs coordinate preferably with the exocyclic carbon atom to Mg
2+ or Zn
2+ (η
1 bonding). This is in accordance with experimental findings; end-on rather than side-on coordination is typical for NHOs and has been described for a range of different metal complexes [
13,
17,
18,
33]. Hence, the parameters
d1 and
d2, defined as the distances of the metal to the exo- and endocyclic olefinic carbon (
Scheme 2), can serve as a direct probe for any degree of side-on coordination. Interestingly, a direct comparison immediately reveals differences in this respect for the magnesium-, zinc- and lithium-complexes. While the MgCl
2 adduct (
Table 1) displays a clear preference for end-on binding, this property is even more pronounced for the ZnCl
2-based analogue (
Table 2), where the corresponding Δ
d is largest although the average bonding distances
d1 and
d2 are even somewhat smaller than found for the series of MgCl
2-complexes. In contrast, the LiCl series displays larger
d values, implying a rather weak coordination. Also, the asymmetry between
d1 and
d2 is milder and in some cases the lithium takes up a virtually equidistant position relative to the two olefinic carbons. This effect is obviously influenced by the chemical structure of the NHO and most pronounced for
4, which may be explained by the relatively low double bond-polarization in this molecule (see below). The increasing tendency to form end-on coordinated adducts in the sequence Li
+ < Mg
2+ < Zn
2+ as described by
d1/
d2 is coherently mirrored by the Gibbs Free Energy of the reaction, which shows the adduct formation to be increasingly exothermic in the same sequence.
This is true for the full range of all eight investigated NHOs, suggesting that for any given system the proportion of free NHO is highest for LiCl and lowest for ZnCl
2 (for the latter, the reaction enthalpy is by 15 kJ/mol smaller on average than found for MgCl
2). The stronger tendency of ZnCl
2 than MgCl
2 to bind to the investigated NHOs is also expressed in shorter Zn-C bond lengths (compare
Table 1 and
Table 2, on average 0.15 Å shorter), in spite of the very similar ionic radii of Mg
2+ and Zn
2+. The binding to Li
+ is unfavorable compared to the binding affinity to Mg
2+ by up to 45 kJ/mol and compared to ZnCl
2 by up to 65 kJ/mol, depending on the NHO. In this context it should be noted that the same tendency seems to apply for analogous NHC complexes (MgCl
2/ZnCl
2) [
4], supporting the findings in this work.
The chemical structure of the NHO significantly influences the reaction energies, allowing for some instructive comparisons. The results are more clearly cut for the zinc- and magnesium analogues—not surprisingly, since here a proper bond is formed—while for the LiCl complexes the weak interaction equalizes the reaction energies. Thus, for compounds
1–
3, a series of NHOs with no further substituent on the exocyclic carbon (C=CH
2 moiety), the bonding propensity increases on going from
1 over
2 to
3. This nicely matches literature reports, which indicate that a six-membered, tetrahydropyrimidine-based structure such as
2 should be somewhat more reactive than
1 [
34], while imidazole derivatives (
3) display a striking reactivity on account of their extreme double bond polarization (in C
6D
6,
3 shows a shift of δ = 2.77 ppm for its olefinic protons [
21]). This behavior can be explained by the formation of an aromatic imidazolium moiety once charge separation occurs for NHOs such as
3 or
6 (see also
Figure 1b). Reports consequently describe a very different behavior in organopolymerization, where imidazole-based NHOs frequently are able to polymerize while the saturated analogues remain inactive, as was found for lactones, epoxides or acrylic monomers [
14,
31,
35]. The increased electron density on the exocyclic carbon should therefore render NHO
3 most prone for coordination to a Lewis acid, a conclusion that is supported by the data listed in
Table 1 and
Table 2; the ZnCl
2(
3)(THF) adduct accounts for the most exothermic reaction energy for all investigated NHO/metal halide combinations (−53.4 kJ/mol). The series
4–
6, analogous to
1–
3 but with additional dimethyl-substitution on the exocyclic carbon (C=C(CH
3)
2 moiety) is of significance since this modification is a prerequisite for successful polymerization in lactone or even acrylate polymerization [
31,
35]. This also shows in the binding propensity, which is in all three cases much less pronounced than for the non-substituted compounds. For
4 and
5 (MgCl
2, ZnCl
2) the reaction energies even turn endothermic, while for the imidazole-derivative
6 still a negative free reaction energy was calculated. Obviously, the increased steric congestion disfavors adduct formation (see
Table 4 for buried volumes, %
VBur, of NHOs
1–
8) [
36,
37]. Since compounds
4–
6 have been highly successful in the Lewis pair polymerization of lactones [
11,
12], including the polymerization of VL, CL and PDL as well as the copolymerization of GBL, this destabilization of adducts seems to be beneficial for catalytic activity. Moving to compound
7, a benzimidazole derivative, the data from MgCl
2 and ZnCl
2 coordination put this NHO in an intermediate range between the less polarized saturated ring systems and the imidazole structure
6. This suggests that benzimidazole backbones might be suitable to realize polymerization systems where the NHO is still considerably active, but does not form too strongly associated Lewis pairs. Experimental data for this NHO is scarce, but a report on VL/CL homo- and copolymerization (in cooperation with Lewis acids such as MgCl
2, ZnCl
2, YCl
3) attests to a high catalytic performance with good control over polydispersity (
ÐM = 1.1) [
11]. Finally, NHO
8 shall be considered. For this saturated, five-membered compound a seemingly small change (connecting the exocyclic methyl substituents to form a cyclopropane) can be observed to have a surprising impact. Comparing the reaction energies for
1,
4, and
8 (ZnCl
2) of −26.6 kJ/mol, +7.6 kJ/mol and −44.1 kJ/mol, respectively, reveals that the bonding propensity for this particular NHO is the strongest in this series and even more pronounced than for imidazole derivative
6 (for MgCl
2 the same applies). This is striking; potentially, the electron donating effect exerted by the cyclopropane moiety overcompensates for steric crowding (according to
Table 4 the steric demand of
8 is intermediate between NHOs
1 and
4) and for having a saturated backbone in the heterocyclic ring.
13C-NMR investigations of this compound attest to the high electron density on the exocyclic, olefinic carbon [
20], which can be interpreted to support the calculated propensity for complex formation. Information on the polymerization activity of this NHO does currently not exist, but a high activity in organopolymerization might be expected on the basis of these findings.
Overall, the results described in this section indicate that adduct formation for NHO/metal halides Lewis pairs can be readily influenced—perhaps even tailored—by suitable selection of both components. For example, the 4/LiCl LP can be expected to offer a high degree of “free” cocatalysts, while the application of 3/ZnCl2 should provide a result from the other end of the scale. Furthermore, it was found that the exocyclic substituents are a prime tuning site for regulating adduct formation. The good congruency of experimental findings with the tendencies described in this section also suggest that future NHO (co)catalysts could be conveniently screened in this manner, with a limited computational effort (see Computational Details).
2.2. Coordination of Lactones to MgCl2
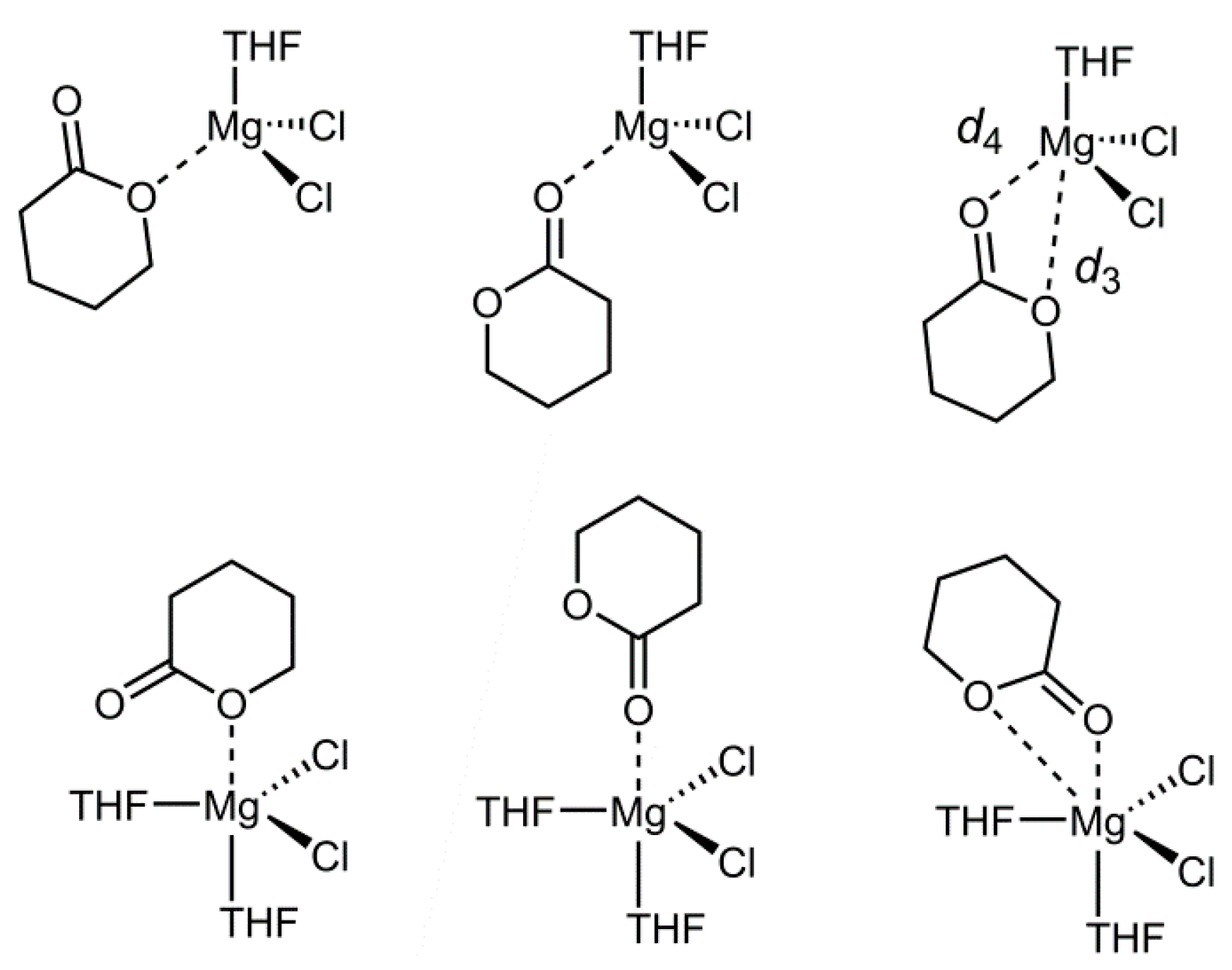
Another central assumption in the discussed type of LP lactone polymerization is the activation of the monomer by the Lewis acid. As a representative example we have chosen MgCl
2 and studied its interaction with β-butyrolactone (BBL), GBL, VL and CL in different coordination geometries and activation modes (
Figure 2). The coordination geometries include (distorted) tetrahedral and trigonal-bipyramidal configurations, while the activation modes include lactone-metal interactions via the endocyclic oxygen, via the carbonyl oxygen and a chelating binding motif employing both oxygens. By choice of the authors, octahedral complexes were not considered; the similar, although not identical, situation with NHC-MgCl
2 complexes suggested low coordination numbers [
4]. For all models it was assumed that the Mg-Cl bonds remain intact and free coordination space is taken up by THF (one solvent molecule for the tetrahedral coordination, two THF molecules for trigonal-bipyramidal configuration). Interaction of the lactone with MgCl
2 in the pentacoordinate scenario always resulted in the apical positioning of the monomer being preferred.
For the optimized structures, results are given in
Table 5 and
Table 6. The distances of Mg to the relevant oxygen atoms are listed, whereby
d3 and
d4 describe the distance to the endocyclic and carbonyl oxygen, respectively. For VL and CL, no minimum structure for the endocyclic binding motif could be found in case of the tetrahedral [MgCl
2(Lactone)(THF)] complex. All attempts to identify this local minimum resulted in the bidentate structure. This implies, before any quantitative evaluation is considered, that the endocyclic type of activation is energetically disfavored. Likewise, no minimum structure for this coordination was obtained for VL and GBL in the trigonal-bipyramidal configuration, i.e., [MgCl
2(Lactone)(THF)
2].
Noticeably, the distances between magnesium and the oxygen atoms are slightly longer for the four-membered BBL, compared to other lactones. This suggests weaker binding to Mg
2+, an assumption that is also mirrored by the corresponding Δ
GR values (see below). Generally, for all lactones the O–Mg distances are larger in the trigonal-bipyramidal coordination, which most probably reflects the increased steric hindrance caused by the fifth ligand. Interestingly, also a preference for certain binding motifs becomes apparent. Coordination via the endocyclic oxygen alone is clearly disfavored. Even where such species were identified,
d3 is relatively long with 2.16–2.17 Å (tetrahedral) and 2.39–2.64 Å (trigonal-bipyramidal). For comparison, the bond length to THF in these complexes is 2.08 Å and 2.10/2.20 (equatorial/apical), respectively. This implies a rather weak bonding, which is potentially caused by the reduced electron density on this oxygen atom compared to its carbonyl counterpart and the steric demand exerted by the neighboring carbonyl group, impairing coordination (see bottom left structure,
Figure 2). For the bidentate activation mode, only rather high
d3 values were found (2.79–3.45 Å, 3.10–3.47 Å), while the distance to the carbonyl oxygen (
d4) was in the range of 2.09–2.10 Å and 2.17–2.22 Å, respectively, for the two geometries investigated. While this negates a truly chelating coordination of the lactone, it is nonetheless interesting to note that the Mg–O (
d3) interaction significantly grows upon increasing the ring size of the monomer, as evidenced by the shrinking interatomic distances reported in
Table 5 and
Table 6.
The most probable explanation for this behavior is found in the required four-membered cycle (Mg–O–C–O), which is clearly strained. However, this strain lessens when the O–C–O angle gets smaller; consequently, for BBL with its rigid structure and largest O-C-O angle an effective bidentate activation is clearly disfavored, while CL with its smaller corresponding angle and higher flexibility can achieve a distance
d3 of 2.79 Å, indicating a weak interaction. The third mode of activation, exclusively via the carbonyl oxygen, delivers the shortest Mg-O distances of all investigated binding motifs, suggesting it as the preferred coordination type for the lactone. This is also supported by another indicator for the strength of the binding of the lactones to magnesium, namely the binding energy derived from the Gibbs Free Energy of the reaction [
39,
40,
41]:
Geometries as depicted in
Figure 2 were employed. The results of these calculations are summarized in
Table 7.
The Gibbs Free Energy of the reaction for BBL is generally higher than the corresponding data of the other lactones, implying that BBL is less likely bound and thereby also less likely to be activated for ring-opening in this manner. Albeit circumstantial, it is still noteworthy that so far no reports for BBL polymerization using this type of LP exist, perhaps a consequence of the seemingly ineffectual activation.
For all lactones, the endocyclic binding mode displays a rather large average reaction energy of +18.3 kJ/mol. Together with the findings on Mg–O bond lengths (see above) this suggests that this mode of coordination can be considered less likely. For the bidentate binding mode, the average reaction energy (+5.3 kJ/mol) is slightly higher than the average reaction energy of the carbonyl-only motif (+3.6 kJ/mol), thus the latter type of activation is again rendered the most favored one. Hence, overall an endocylic-only coordination can be discounted, while activation via the carbonyl is most likely. Tentatively, this can be correlated with experimental observations. A YCl
3(CL)
3 complex has been characterized by crystal data analysis [
42], whereby all three CL ligands in this complex are exclusively coordinated via the carbonyl oxygen. However, it is unclear whether the same applies in solution. A chelate-like coordination by the lactone cannot be ruled out by the results discussed above. The stepwise increasing ability of the lactone to achieve coordination via both oxygen species as the ring size grows from the four-membered BBL to the seven-membered CL (especially tetrahedral coordination, see
Table 5) could indicate that this is one of the mechanisms by which monomer selectivity is engendered in the NHO/metal halide LP polymerization setup.
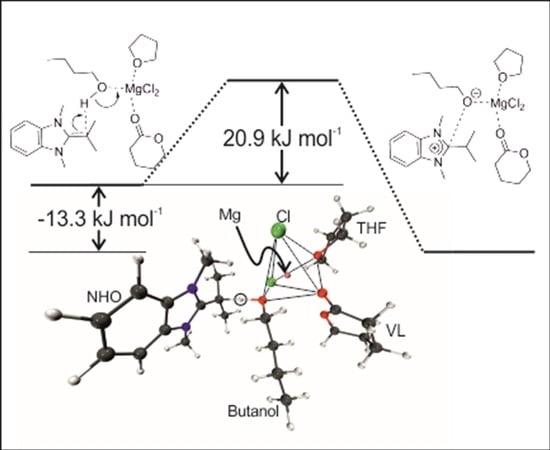
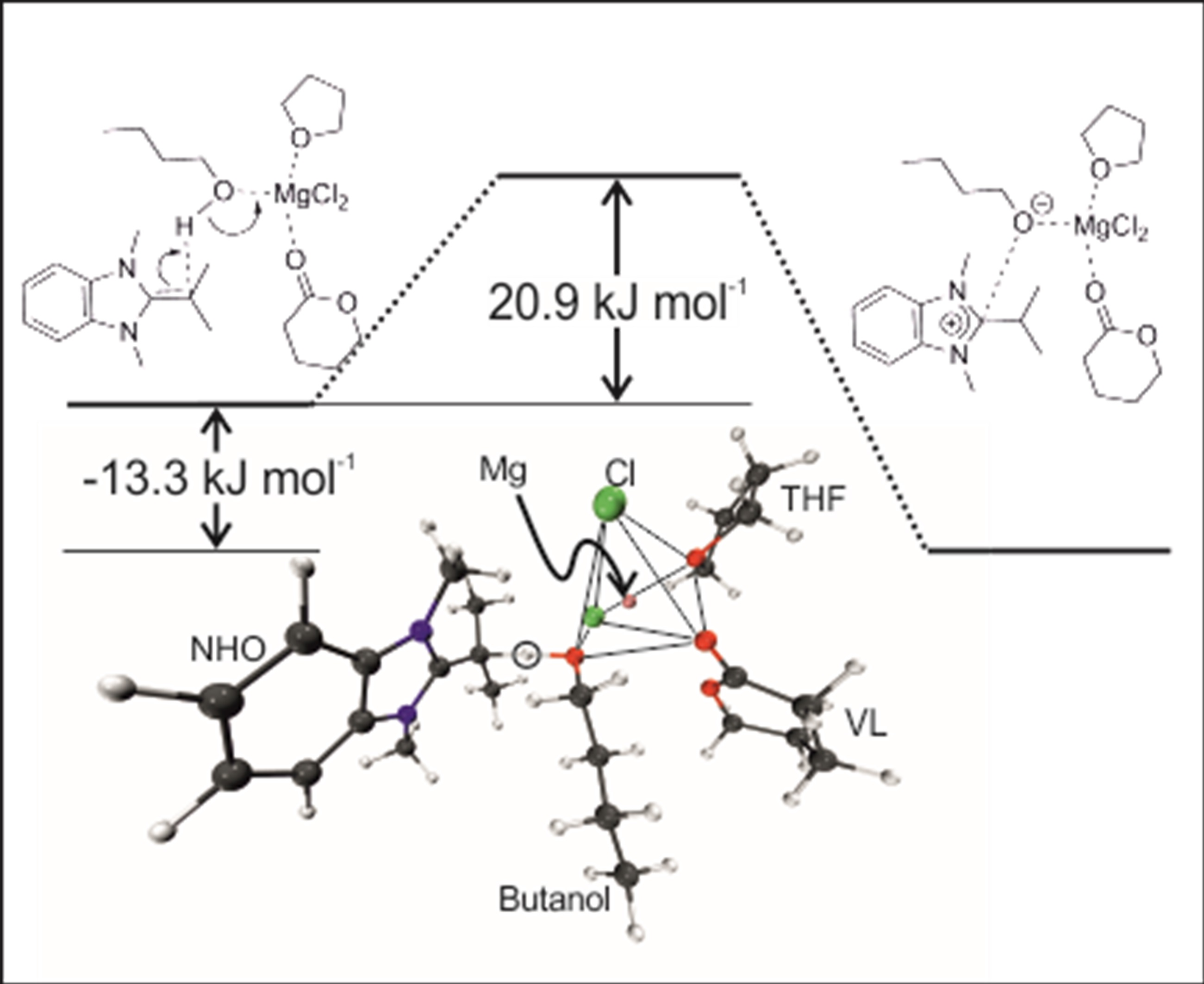
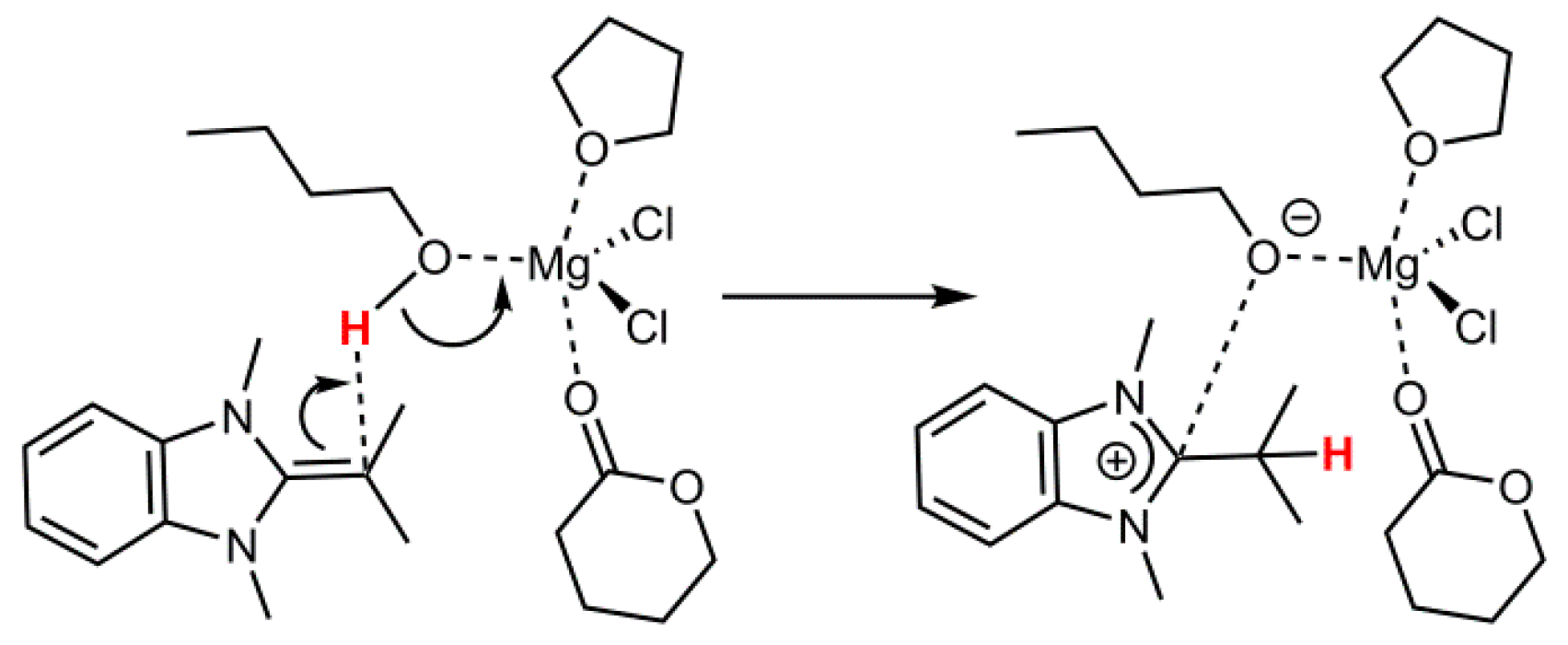
2.3. Initiation: Proton Transfer Step
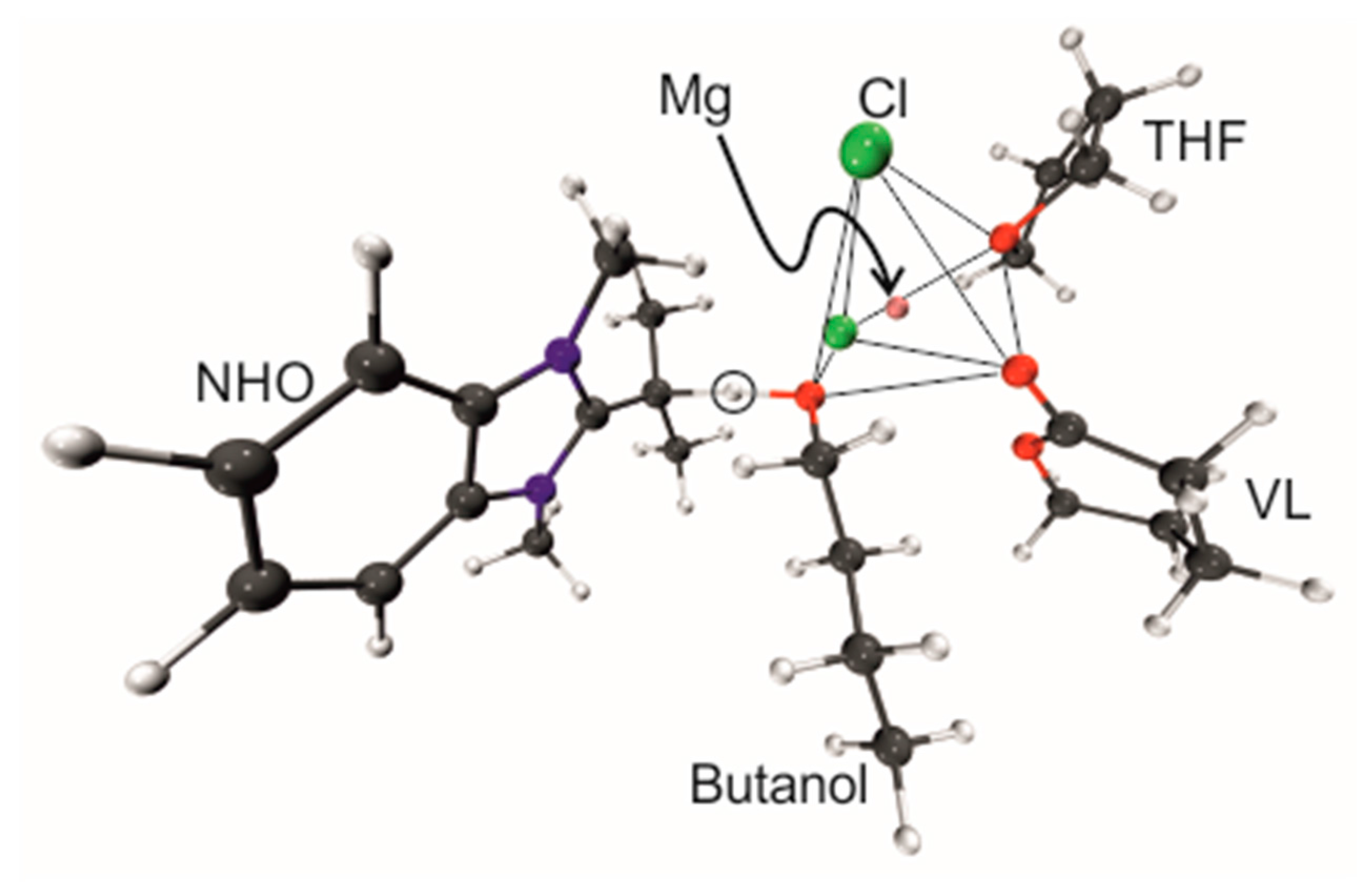
Finally, the initial step of the polymerization process, which is often described as the deprotonation/activation of an initiating alcohol species, was also investigated. As model components, the interaction of MgCl
2 with one VL and one THF molecule in the presence of butanol was investigated. As NHO compound
7 was selected (
Scheme 3,
Figure 3), since this structure represents intermediate polarization. Also, the successful polymerization of VL by
7/MgCl
2 has been reported in the presence of an alcohol as initiator (conversion > 90%, 80 min, 1% NHO-loading,
ÐM = 1.10–1.20) [
11].
Calculations revealed a potential activation energy barrier of 30.2 kJ/mol for the proton transfer (20.9 kJ/mol including zero point energy (ZPE)) and a potential reaction energy of −14.5 kJ/mol (−13.3 kJ/mol including ZPE), rendering this process well feasible. However,
without MgCl
2 being present, the deprotonation of butanol with NHO
7 was calculated to be endothermic by approximately 65 kJ/mol (see also
Figure S1, Supplementary Materials). Significantly, this is in accordance with recent experimental descriptions of the deprotonation of benzyl alcohol, a typical initiator for lactone polymerization, where it was detailed that in the absence of metal-based Lewis acids no proton transfer was observed (
1H-NMR), yet with different metal halides varying proportions of protonated NHO species were detected [
12]. Thus, especially less reactive NHOs (such as
1,
2,
4,
5 or
7) seem to require the additional support by a Lewis acid to efficiently polymerize lactones, most probably because the electron deficient metal species stabilizes the formation of an oxyanionic species (butanolate) from the initiator and thus acidifies the –OH functional group.
During the course of the proton transfer the coordination sphere of Mg
2+ remains broadly unchanged. The Mg-O
butanol distance (
Table 8) shrinks from 2.13 Å to 1.95 Å, nicely mirroring the stronger bonding of the butanolate to Mg
2+ (as compared to the initial butanol). The olefinic C=C double bond in the NHO is converted into a single bond upon protonation, which is reflected by the changing C–C distance, growing from 1.39 Å to 1.50 Å.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}