The Effects of Early-Onset Pre-Eclampsia on Placental Creatine Metabolism in the Third Trimester

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Population Characteristics
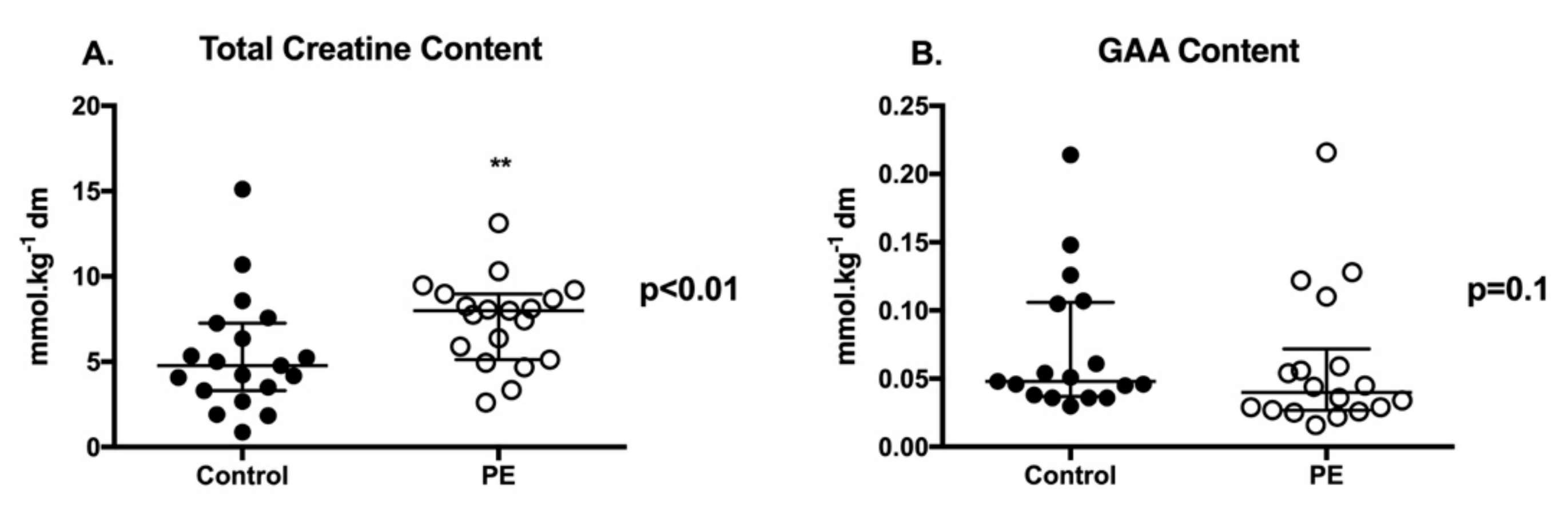
2.2. Placental Creatine and GAA Content
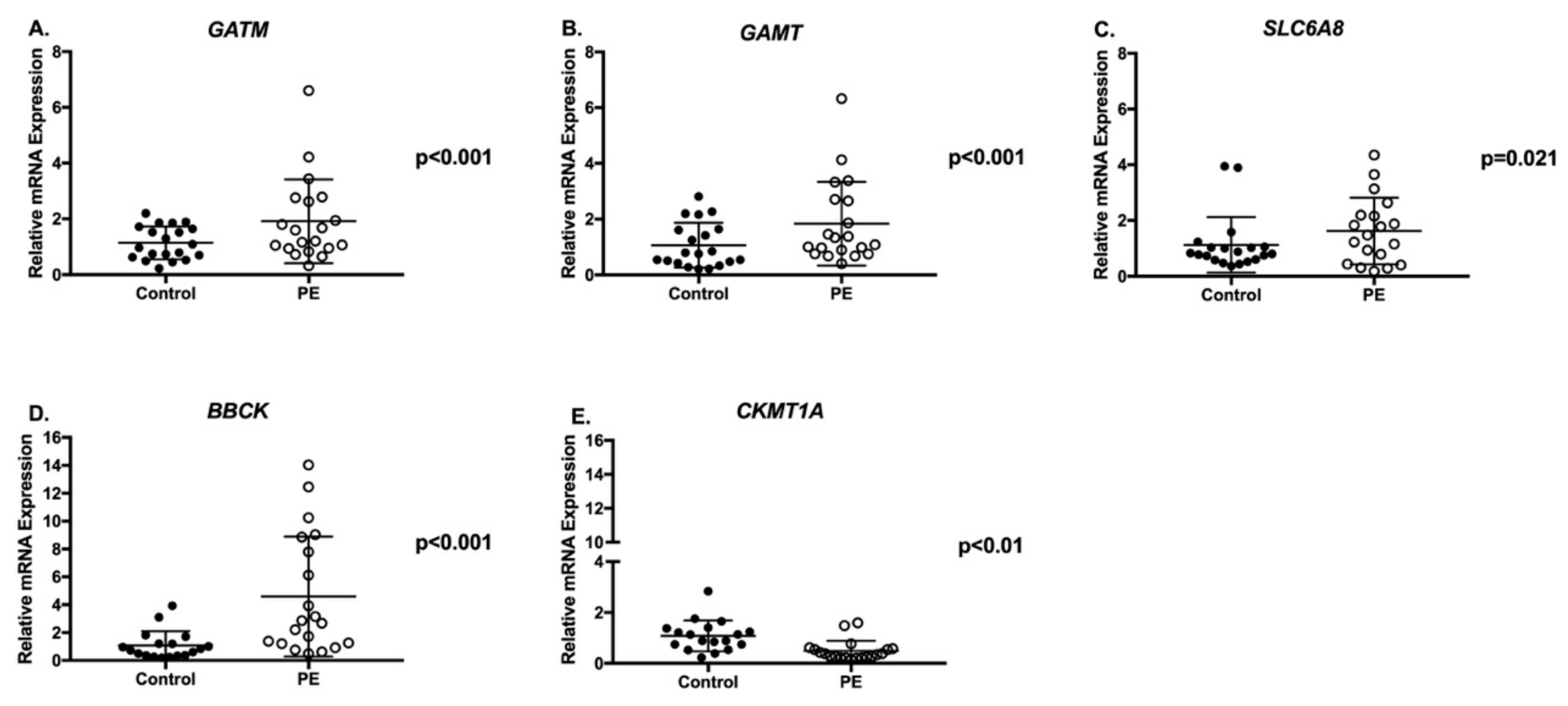
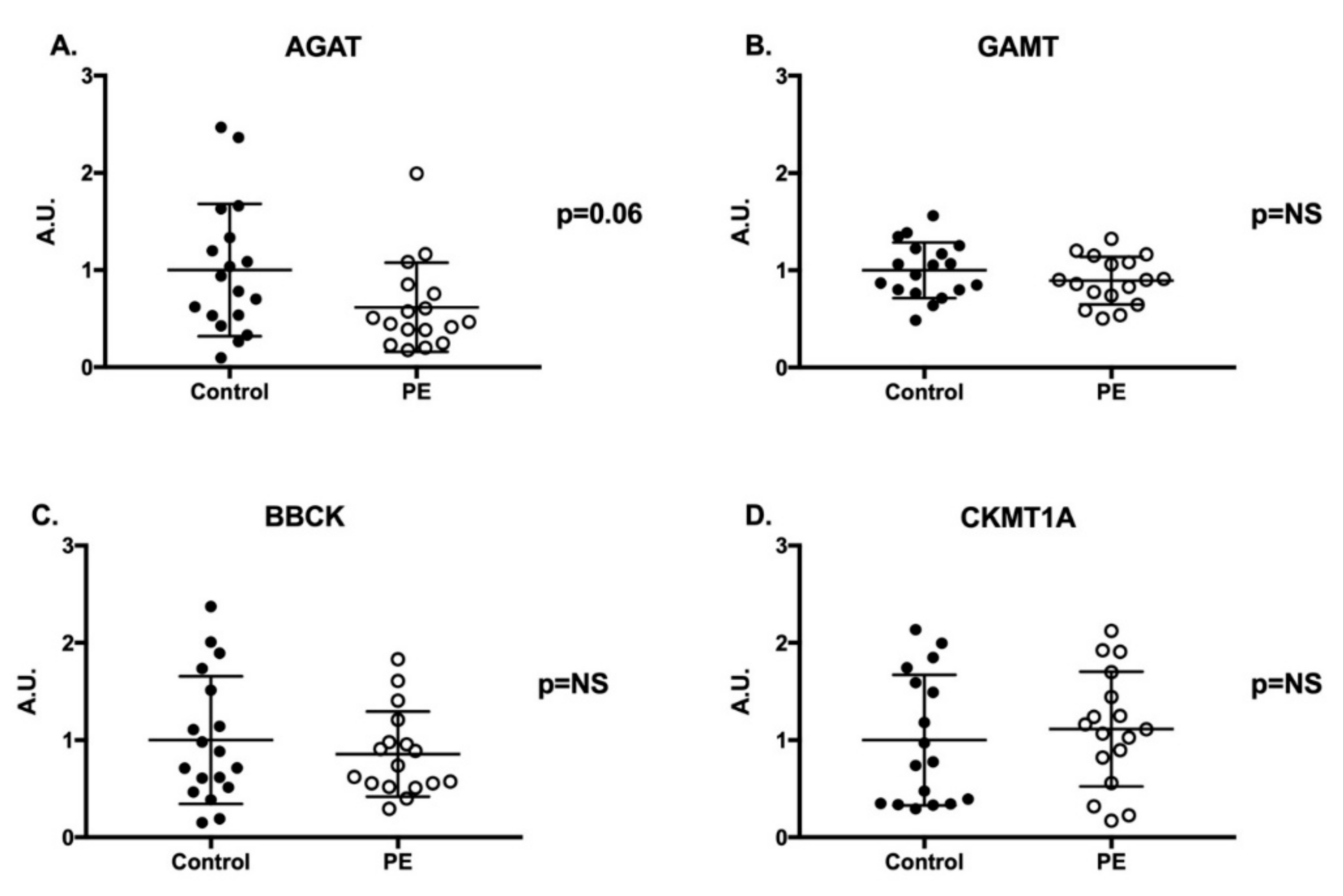
2.3. Creatine Synthesis and Transport Genes and Proteins
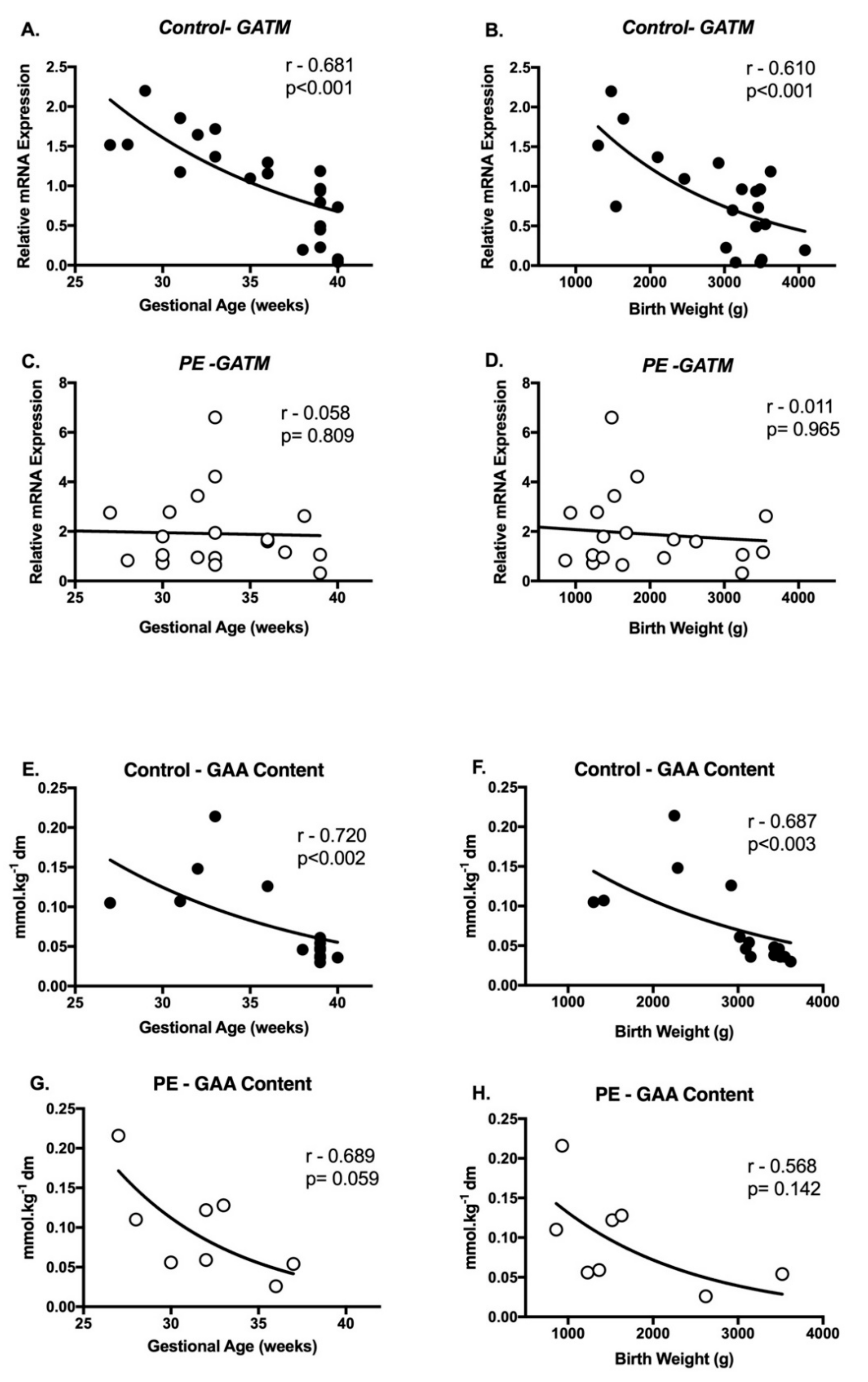
2.4. Correlations between Creatine Metabolism and Pregnancy Outcomes
3. Discussion
4. Materials and Methods
4.1. Human Research Ethics, Sample Collection and Storage
4.2. Sample Processing
4.2.1. Creatine and GAA Analysis
4.2.2. Gene Expression Analysis
4.2.3. Protein Analysis
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PE | Pre-eclampsia |
| GAA | Guanidinoacetate |
| GATM | Gene—arginine:glycine aminotransferase |
| AGAT | Protein—arginine:glycine aminotransferase |
| SLC6A8 | Gene—creatine transporter |
| GAMT | Guanidinoacetate methyltransferas |
| BBCK | Cytosolic creatine kinase |
| CKMT1A | Mitochondrial creatine kinase |
| ADP | Adenosine Diphosphate |
| ATP | Adenosine Triphosphate |
| I/R | Ischemic-reperfusion |
| FGR | Fetal Growth Restriction |
| mRNA | Messenger Ribonucleic Acid |
| AMPK | AMP-activated protein kinase |
| PGC-1α | Peroxisome activated receptor γ coactivator-1 alpha |
| ERRα | Estrogen-related receptor α |
| HIF-2α | Hypoxia-inducible factor 2 α |
References
- Ananth, C.V.; Keyes, K.M.; Wapner, R.J. Pre-eclampsia rates in the United States, 1980–2010: Age-period-cohort analysis. BMJ 2013, 347, f6564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mol, B.W.; Roberts, C.T.; Thangaratinam, S.; Magee, L.A.; De Groot, C.J.; Hofmeyr, G.J. Pre-eclampsia. Lancet 2016, 387, 999–1011. [Google Scholar] [CrossRef]
- Abalos, E.; Cuesta, C.; Grosso, A.L.; Chou, D.; Say, L. Global and regional estimates of preeclampsia and eclampsia: A systematic review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 170, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ghulmiyyah, L.; Sibai, B. Maternal mortality from preeclampsia/eclampsia. Semin. Perinatol. 2012, 36, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, J.E.; Fraser, R.; Leslie, K.; Wallace, A.E.; James, J.L. Remodelling at the maternal-fetal interface: Relevance to human pregnancy disorders. Reproduction 2010, 140, 803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karumanchi, S.A. Angiogenic factors in preeclampsia: From diagnosis to therapy. Hypertension 2016, 67, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.W.; Sacks, G.P.; Sargent, I.L. Preeclampsia: An excessive maternal inflammatory response to pregnancy. Am. J. Obstet. Gynecol. 1999, 180, 499–506. [Google Scholar] [CrossRef]
- Holland, O.; Nitert, M.D.; Gallo, L.A.; Vejzovic, M.; Fisher, J.J.; Perkins, A.V. Placental mitochondrial function and structure in gestational disorders. Placenta 2017, 54, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Jääskeläinen, T.; Kärkkäinen, O.; Jokkala, J.; Litonius, K.; Heinonen, S.; Auriola, S.; Lehtonen, M.; Hanhineva, K.; Laivuori, H. A non-targeted LC-MS profiling reveals elevated levels of carnitine precursors and trimethylated compounds in the cord plasma of pre-eclamptic infants. Sci. Rep. 2018, 8, 14616. [Google Scholar] [CrossRef]
- Ellery, S.J.; Della Gatta, P.A.; Bruce, C.R.; Kowalski, G.M.; Davies-Tuck, M.; Mockler, J.C.; Murthi, P.; Walker, D.W.; Snow, R.J.; Dickinson, H. Creatine biosynthesis and transport by the term human placenta. Placenta 2017, 52, 86–93. [Google Scholar] [CrossRef]
- Wallimann, T.; Tokarska-Schlattner, M.; Schlattner, U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 2011, 40, 1271–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braissant, O.; Henry, H.; Villard, A.-M.; Speer, O.; Wallimann, T.; Bachmann, C. Creatine synthesis and transport during rat embryogenesis: Spatiotemporal expression of AGAT, GAMT and CT1. BMC Dev. Biol. 2005, 5, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomure, M. Regulation of creatine kinase isoenzymes in human placenta during early, mid-, and late gestation. J. Soc. Gynaecol. Investig. 1996, 3, 322–327. [Google Scholar] [CrossRef]
- Dickinson, H.; Davies-Tuck, M.; Ellery, S.; Grieger, J.; Wallace, E.; Snow, R.; Walker, D.; Clifton, V. Maternal creatine in pregnancy: A retrospective cohort study. Bjog: Int. J. Obstet. Gynaecol. 2016, 123, 1830–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heazell, A.E.; Bernatavicius, G.; Warrander, L.; Brown, M.C.; Dunn, W.B. A metabolomic approach identifies differences in maternal serum in third trimester pregnancies that end in poor perinatal outcome. Reprod. Sci. 2012, 19, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, I.E.; Du Plessis, A.J.; Vezina, G.; Noeske, R.; Limperopoulos, C. Elucidating metabolic maturation in the healthy fetal brain using 1H-MR spectroscopy. Am. J. Neuroradiol. 2016, 37, 360–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellery, S.J.; Murthi, P.; Davies-Tuck, M.L.; Gatta, P.D.; May, A.K.; Kowalski, G.M.; Callahan, D.L.; Bruce, C.R.; Alers, N.O.; Miller, S.L. Placental Creatine Metabolism in Cases of Placental Insufficiency and Reduced Fetal Growth. Mol. Hum. Reprod. 2019, 25, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Redman, C.W.; Roberts, J.M.; Moffett, A. Pre-eclampsia: Pathophysiology and clinical implications. BMJ 2019, 366, l2381. [Google Scholar] [CrossRef] [Green Version]
- Tissot van Patot, M.C.; Murray, A.J.; Beckey, V.; Cindrova-Davies, T.; Johns, J.; Zwerdlinger, L.; Jauniaux, E.; Burton, G.J.; Serkova, N.J. Human placental metabolic adaptation to chronic hypoxia, high altitude: Hypoxic preconditioning. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 298, R166–R172. [Google Scholar] [CrossRef] [Green Version]
- Brosnan, J.; Brosnan, M. Creatine: Endogenous metabolite, dietary, and therapeutic supplement. Annu. Rev. Nutr. 2007, 27, 241–261. [Google Scholar] [CrossRef] [Green Version]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and creatinine metabolism. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef] [PubMed]
- Edison, E.E.; Brosnan, M.E.; Meyer, C.; Brosnan, J.T. Creatine synthesis: Production of guanidinoacetate by the rat and human kidney in vivo. Am. J. Physiol. Ren. Physiol. 2007, 293, F1799–F1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuire, D.M.; Tormanen, C.; Segal, I.; Van Pilsum, J. The effect of growth hormone and thyroxine on the amount of L-arginine: Glycine amidinotransferase in kidneys of hypophysectomized rats. Purification and some properties of rat kidney transamidinase. J. Biol. Chem. 1980, 255, 1152–1159. [Google Scholar] [PubMed]
- Guthmiller, P.; Van Pilsum, J.; Boen, J.R.; McGuire, D.M. Cloning and sequencing of rat kidney L-arginine: Glycine amidinotransferase. Studies on the mechanism of regulation by growth hormone and creatine. J. Biol. Chem. 1994, 269, 17556. [Google Scholar] [PubMed]
- Mittal, P.; Espinoza, J.; Hassan, S.; Kusanovic, J.P.; Edwin, S.S.; Nien, J.K.; Gotsch, F.; Than, N.G.; Erez, O.; Mazaki-Tovi, S. Placental growth hormone is increased in the maternal and fetal serum of patients with preeclampsia. J. Matern. Fetal Neonatal Med. 2007, 20, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.-w.; Calabrese, S.; Hynx, D.; Hemmings, B.A.; Cetin, I.; Charnock-Jones, D.S.; Burton, G.J. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am. J. Pathol. 2008, 173, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Sankar, K.D.; Bhanu, P.S.; Ramalingam, K.; Kiran, S.; Ramakrishna, B. Histomorphological and morphometrical changes of placental terminal villi of normotensive and preeclamptic mothers. Anat. Cell Biol. 2013, 46, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Snow, R.J.; Murphy, R.M. Creatine and the creatine transporter: A review. Mol. Cell. Biochem. 2001, 224, 169–181. [Google Scholar] [CrossRef]
- Yang, X.; Xu, P.; Zhang, F.; Zhang, L.; Zheng, Y.; Hu, M.; Wang, L.; Han, T.-l.; Peng, C.; Wang, L. AMPK Hyper-Activation Alters Fatty Acids Metabolism and Impairs Invasiveness of Trophoblasts in Preeclampsia. Cell. Physiol. Biochem. 2018, 49, 578–594. [Google Scholar] [CrossRef]
- Darrabie, M.D.; Arciniegas, A.J.L.; Mishra, R.; Bowles, D.E.; Jacobs, D.O.; Santacruz, L. AMPK and substrate availability regulate creatine transport in cultured cardiomyocytes. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E870–E876. [Google Scholar] [CrossRef] [Green Version]
- Huss, J.M.; Torra, I.P.; Staels, B.; Giguere, V.; Kelly, D.P. Estrogen-related receptor α directs peroxisome proliferator-activated receptor α signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell. Biol. 2004, 24, 9079–9091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.L.; Snow, R.J.; Wright, C.R.; Cho, Y.; Wallace, M.A.; Kralli, A.; Russell, A.P. PGC-1α and PGC-1β increase CrT expression and creatine uptake in myotubes via ERRα. Biochim. Biophys. Acta 2014, 1843, 2937–2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishnyakova, P.A.; Volodina, M.A.; Tarasova, N.V.; Marey, M.V.; Tsvirkun, D.V.; Vavina, O.V.; Khodzhaeva, Z.S.; Kan, N.E.; Menon, R.; Vysokikh, M.Y. Mitochondrial role in adaptive response to stress conditions in preeclampsia. Sci. Rep. 2016, 6, 32410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Wang, Z.; Sun, Y. Reduced amount of cytochrome c oxidase subunit I messenger RNA in placentas from pregnancies complicated by preeclampsia. Acta Obstet. Et Gynecol. Scand. 2004, 83, 144–148. [Google Scholar] [CrossRef]
- Schlattner, U.; Klaus, A.; Rios, S.R.; Guzun, R.; Kay, L.; Tokarska-Schlattner, M. Cellular compartmentation of energy metabolism: Creatine kinase microcompartments and recruitment of B-type creatine kinase to specific subcellular sites. Amino Acids 2016, 48, 1751–1774. [Google Scholar] [CrossRef]
- Glover, L.E.; Bowers, B.E.; Saeedi, B.; Ehrentraut, S.F.; Campbell, E.L.; Bayless, A.J.; Dobrinskikh, E.; Kendrick, A.A.; Kelly, C.J.; Burgess, A. Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis. Proc. Natl. Acad. Sci. USA 2013, 110, 19820–19825. [Google Scholar] [CrossRef] [Green Version]
- Holland, O.J.; Cuffe, J.S.; Nitert, M.D.; Callaway, L.; Cheung, K.A.K.; Radenkovic, F.; Perkins, A.V. Placental mitochondrial adaptations in preeclampsia associated with progression to term delivery. Cell Death Dis. 2018, 9, 1150. [Google Scholar] [CrossRef] [Green Version]
- Burton, G.J.; Jauniaux, E. Pathophysiology of placental-derived fetal growth restriction. Am. J. Obstet. Gynecol. 2018, 218, S745–S761. [Google Scholar] [CrossRef] [Green Version]
- Stead, L.M.; Au, K.P.; Jacobs, R.L.; Brosnan, M.E.; Brosnan, J.T. Methylation demand and homocysteine metabolism: Effects of dietary provision of creatine and guanidinoacetate. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E1095–E1100. [Google Scholar] [CrossRef]
- Lai, W.K.C.; Kan, M.Y. Homocysteine-induced endothelial dysfunction. Ann. Nutr. Metab. 2015, 67, 1–12. [Google Scholar] [CrossRef]
- Powers, R.W.; Evans, R.W.; Majors, A.K.; Ojimba, J.I.; Ness, R.B.; Crombleholme, W.R.; Roberts, J.M. Plasma homocysteine concentration is increased in preeclampsia and is associated with evidence of endothelial activation. Am. J. Obstet. Gynecol. 1998, 179, 1605–1611. [Google Scholar] [CrossRef]
- Pidoux, G.; Gerbaud, P.; Laurendeau, I.; Guibourdenche, J.; Bertin, G.; Vidaud, M.; Evain-Brion, D.; Frendo, J.-L. Large variability of trophoblast gene expression within and between human normal term placentae. Placenta 2004, 25, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.; Brown, M.; Dekker, G.; Gatt, S.; McLintock, C.; McMahon, L. Guidelines for the Management of Hypertensive Disorders of Pregnancy; SOMANZ: Sydney, Australia, 2008; pp. 12–23. [Google Scholar]
- Murthi, P.; Said, J.; Doherty, V.; Donath, S.; Nowell, C.; Brennecke, S.; Kalionis, B. Homeobox gene DLX4 expression is increased in idiopathic human fetal growth restriction. Mhr: Basic Sci. Reprod. Med. 2006, 12, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Chui, A.; Murthi, P.; Brennecke, S.; Ignjatovic, V.; Monagle, P.; Said, J.M. The expression of placental proteoglycans in pre-eclampsia. Gynecol. Obstet. Investig. 2012, 73, 277–284. [Google Scholar] [CrossRef]
- Harris, R.C.; Soderlund, K.; Hultman, E. Elevation of creatine in resting and exercised muscle of normal subjects by creatine supplementation. Clin. Sci 1992, 83, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Tran, C.; Yazdanpanah, M.; Kyriakopoulou, L.; Levandovskiy, V.; Zahid, H.; Naufer, A.; Isbrandt, D.; Schulze, A. Stable isotope dilution microquantification of creatine metabolites in plasma, whole blood and dried blood spots for pharmacological studies in mouse models of creatine deficiency. Clin. Chim. Acta 2014, 436, 160–168. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. GenomeBiologycom 2002, 3, 31–34. [Google Scholar]
- Eaton, S.L.; Roche, S.L.; Hurtado, M.L.; Oldknow, K.J.; Farquharson, C.; Gillingwater, T.H.; Wishart, T.M. Total protein analysis as a reliable loading control for quantitative fluorescent Western blotting. PLoS ONE 2013, 8, e72457. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Control (n = 20) | PE (n = 20) | p Value |
|---|---|---|---|
| Maternal Age (years) | 33.5 (5.0) 1 | 28.9 (4.4) 1 | <0.01 |
| Nulliparous | 6.0 (30.0) 2 | 15.0 (75.0) 2 | <0.01 |
| Gestation (weeks) | 34.2 (4.1) 1 | 32.7 (4.0) 1 | NS |
| Systolic Blood Pressure (mmHg) | <140 | 162.3 (11.8) 1 | - |
| Diastolic Blood Pressure (mmHg) | <90 | 102.9 (5.9) 1 | - |
| Mode of Delivery | |||
| Vaginal Delivery | 5.0 (25.0) 2 | 5.0 (25.0) 2 | NS |
| C-Section not in Labor | 14.0 (70.0) 2 | 5.0 (25.0) 2 | <0.01 |
| C-Section in Labor | 1.0 (5.0) 2 | 10.0 (50.0) 2 | <0.01 |
| Birth Weight (g) | 3140 (2762–3620) 3 | 1443 (1155–3520) 3 | <0.05 |
| Placental Weight (g) | 463.5 (402–830) 3 | 380.0 (323–600) 3 | <0.05 |
| Baby Sex (male) | 10 (50.0) 2 | 14 (70.0) 2 | NS |
| Gene of Interest | TaqMan Probes | |
|---|---|---|
| Creatine Synthesis | GATM | Hs00933793_m1 |
| GAMT | Hs00355745_g1 | |
| Creatine Transport | SLC6A8 | Hs00940515_m1 |
| Creatine Kinases | BBCK | Hs00176484_m1 |
| CKMT1A | Hs00179727_m1 | |
| Housekeeping | RN18S1 | Hs03003631_g1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ellery, S.J.; Murthi, P.; Della Gatta, P.A.; May, A.K.; Davies-Tuck, M.L.; Kowalski, G.M.; Callahan, D.L.; Bruce, C.R.; Wallace, E.M.; Walker, D.W.; et al. The Effects of Early-Onset Pre-Eclampsia on Placental Creatine Metabolism in the Third Trimester. Int. J. Mol. Sci. 2020, 21, 806. https://doi.org/10.3390/ijms21030806
Ellery SJ, Murthi P, Della Gatta PA, May AK, Davies-Tuck ML, Kowalski GM, Callahan DL, Bruce CR, Wallace EM, Walker DW, et al. The Effects of Early-Onset Pre-Eclampsia on Placental Creatine Metabolism in the Third Trimester. International Journal of Molecular Sciences. 2020; 21(3):806. https://doi.org/10.3390/ijms21030806
Chicago/Turabian StyleEllery, Stacey J., Padma Murthi, Paul A. Della Gatta, Anthony K. May, Miranda L. Davies-Tuck, Greg M. Kowalski, Damien L. Callahan, Clinton R. Bruce, Euan M. Wallace, David W. Walker, and et al. 2020. "The Effects of Early-Onset Pre-Eclampsia on Placental Creatine Metabolism in the Third Trimester" International Journal of Molecular Sciences 21, no. 3: 806. https://doi.org/10.3390/ijms21030806