A Small Molecule Stabilizer of the MYC G4-Quadruplex Induces Endoplasmic Reticulum Stress, Senescence and Pyroptosis in Multiple Myeloma

 ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
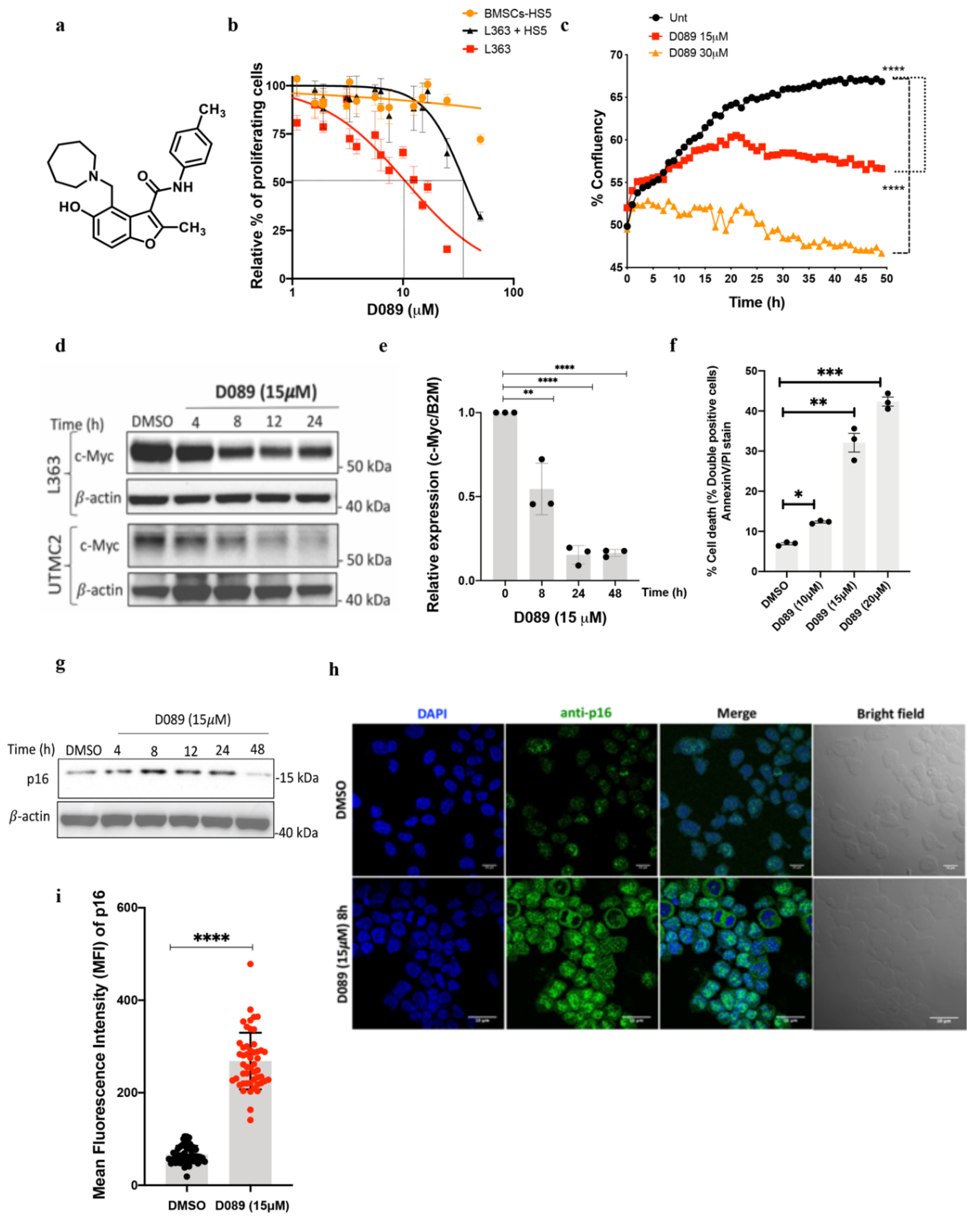
2.1. D089, a Small Molecule Stabilizer of the MYC-G4 Quadruplex Reduces MYC Transcription and Induces Cytotoxicity in Myeloma Cells
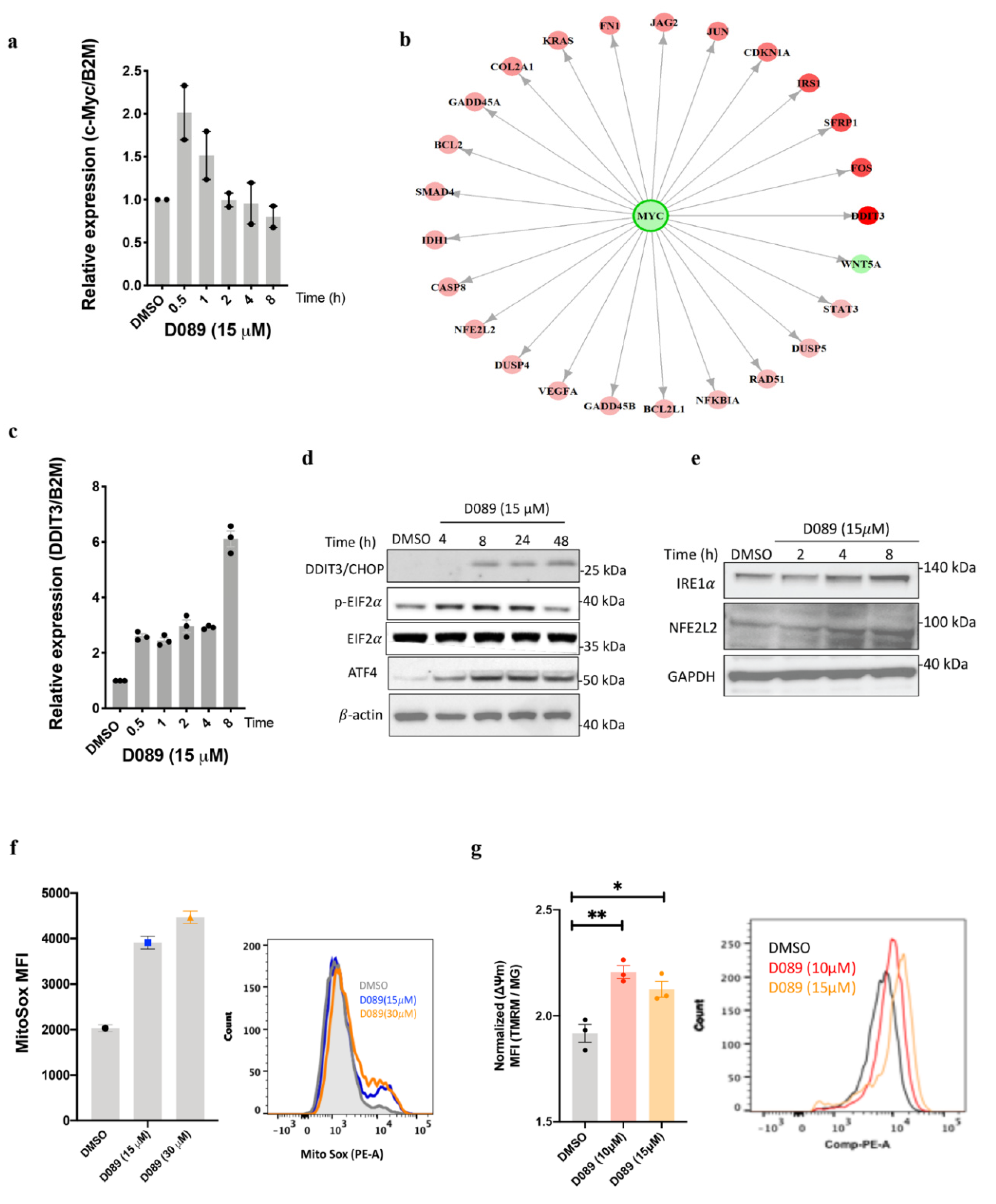
2.2. Drug Treatment Modulates the ER Stress Pathway in Myeloma
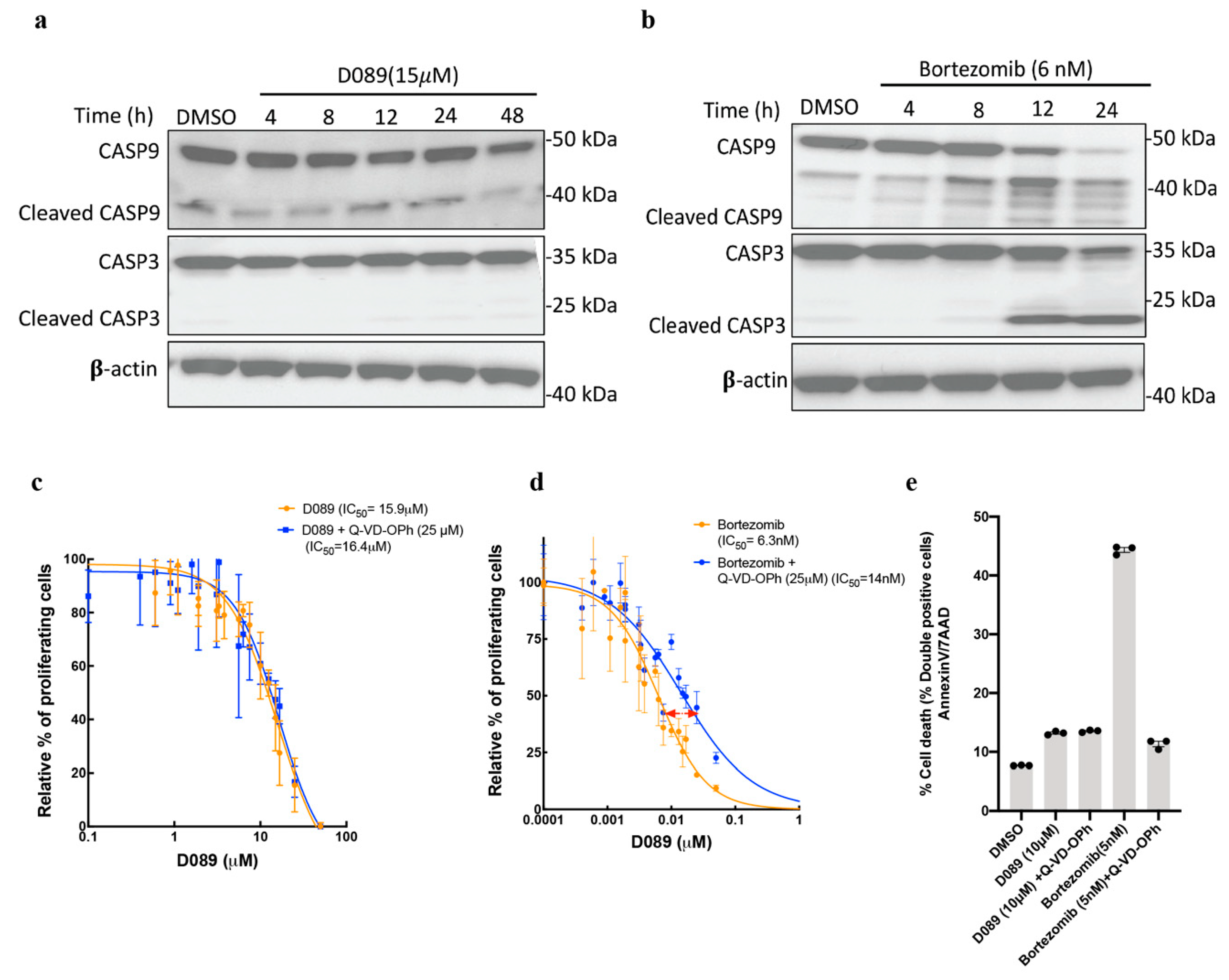
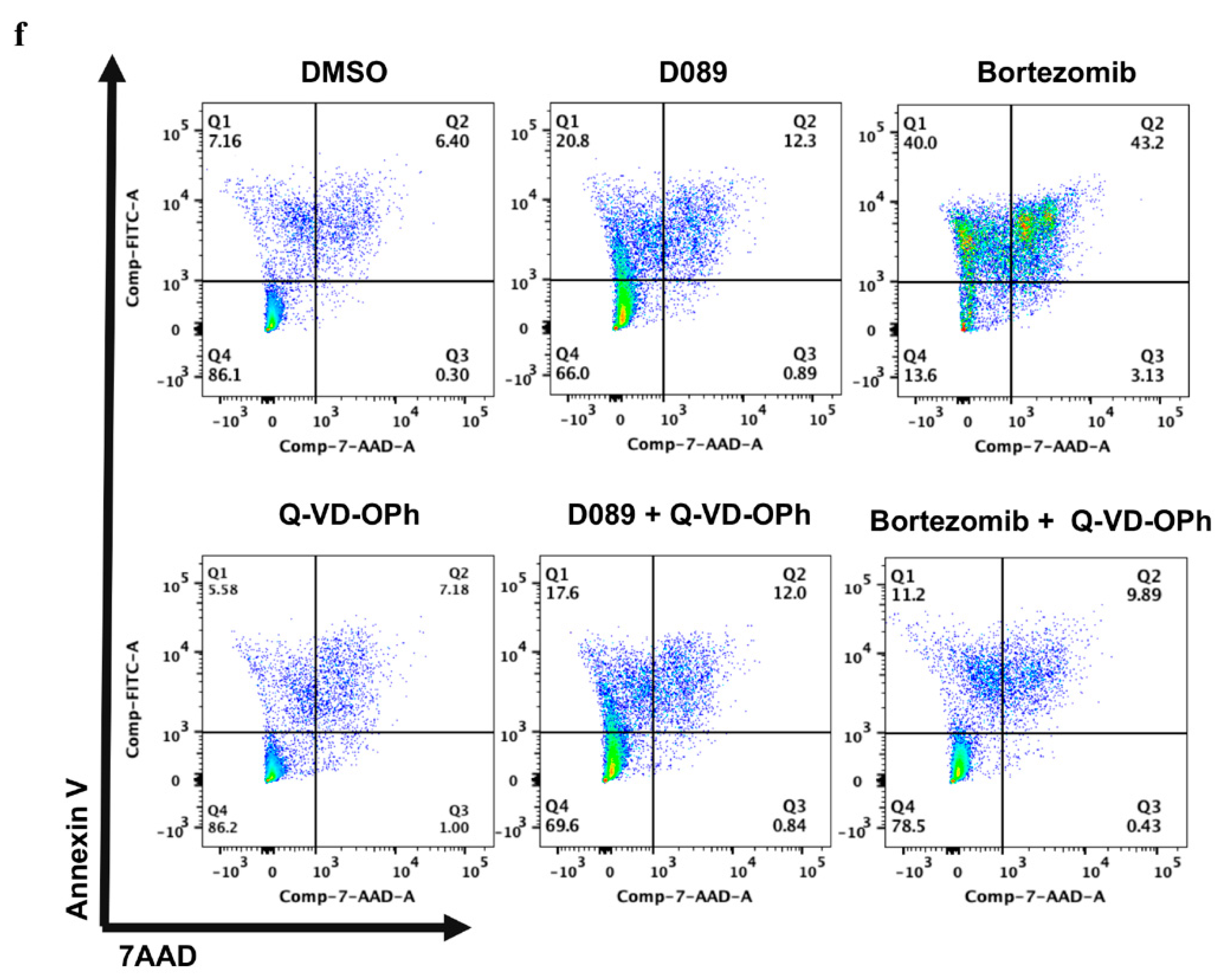
2.3. D089 Cytotoxicity Is Independent of Initiator and Effector Caspases
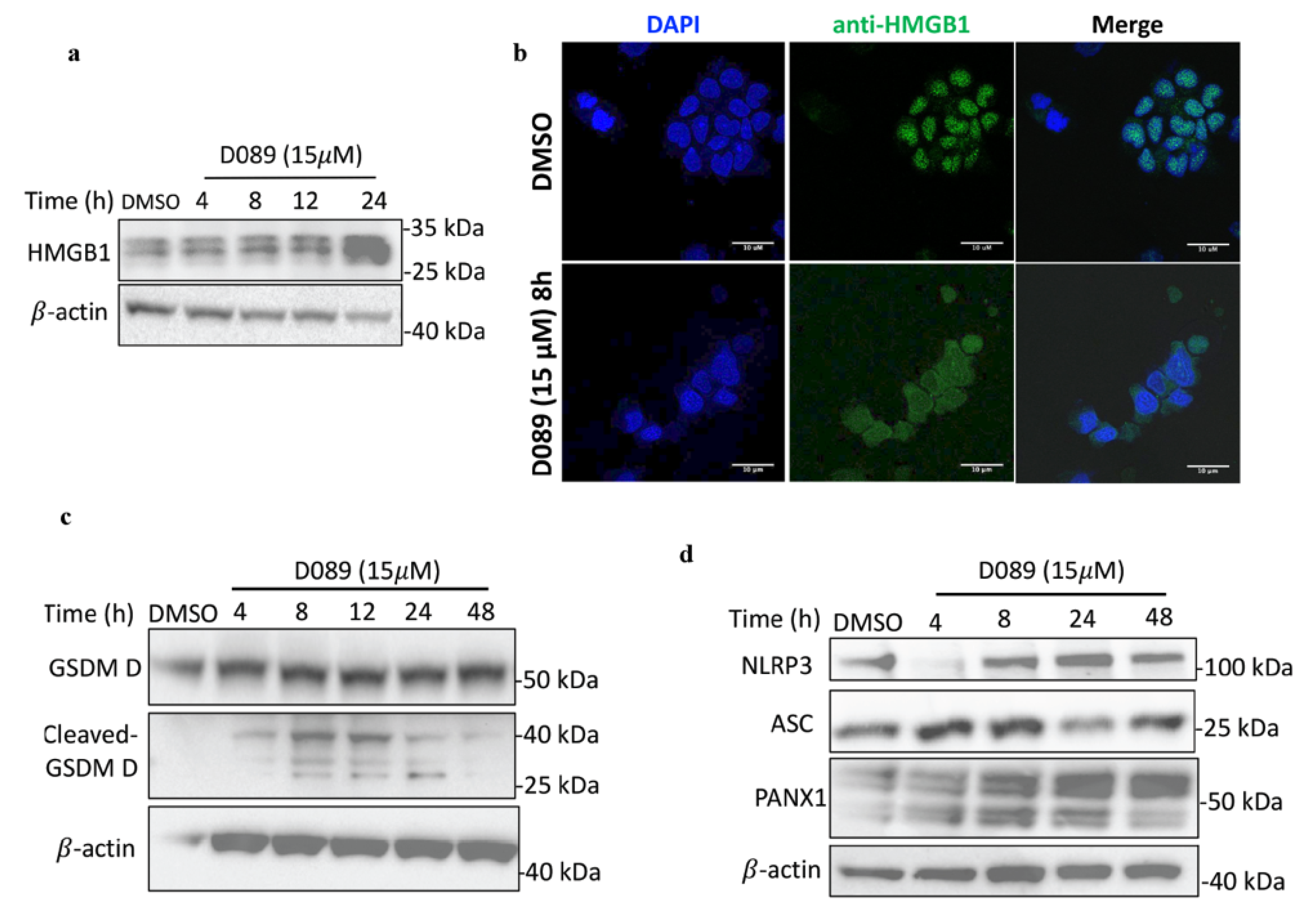
2.4. D089 Induces Caspase-8 and -1 Dependent Cell Death Mechanism-Pyroptosis
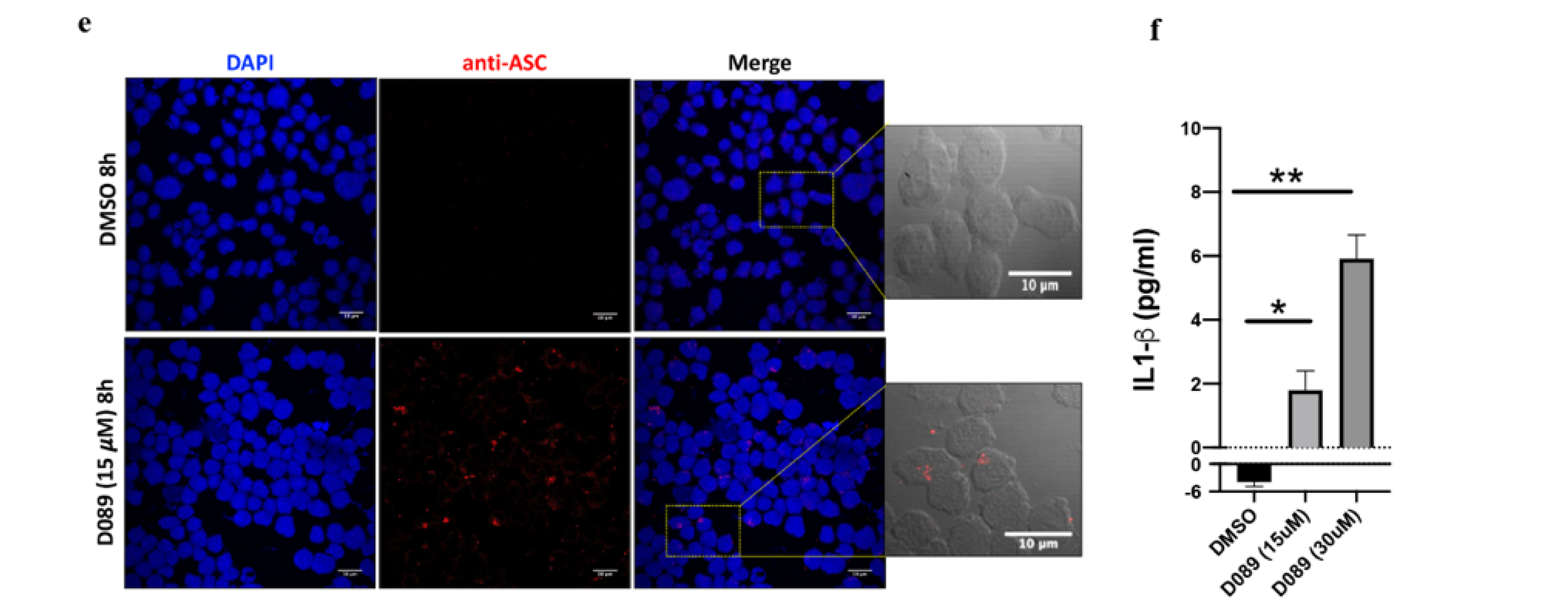
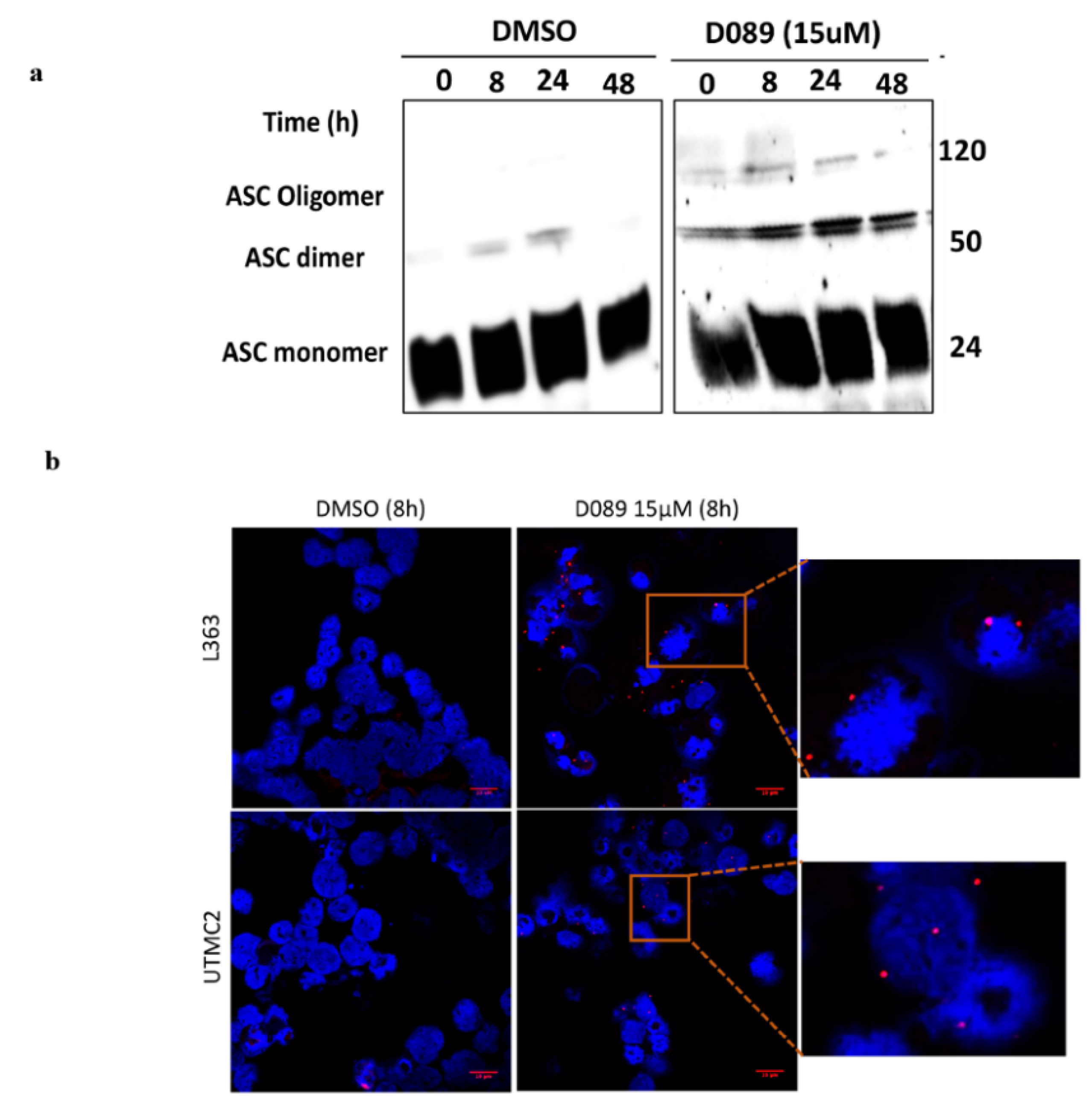
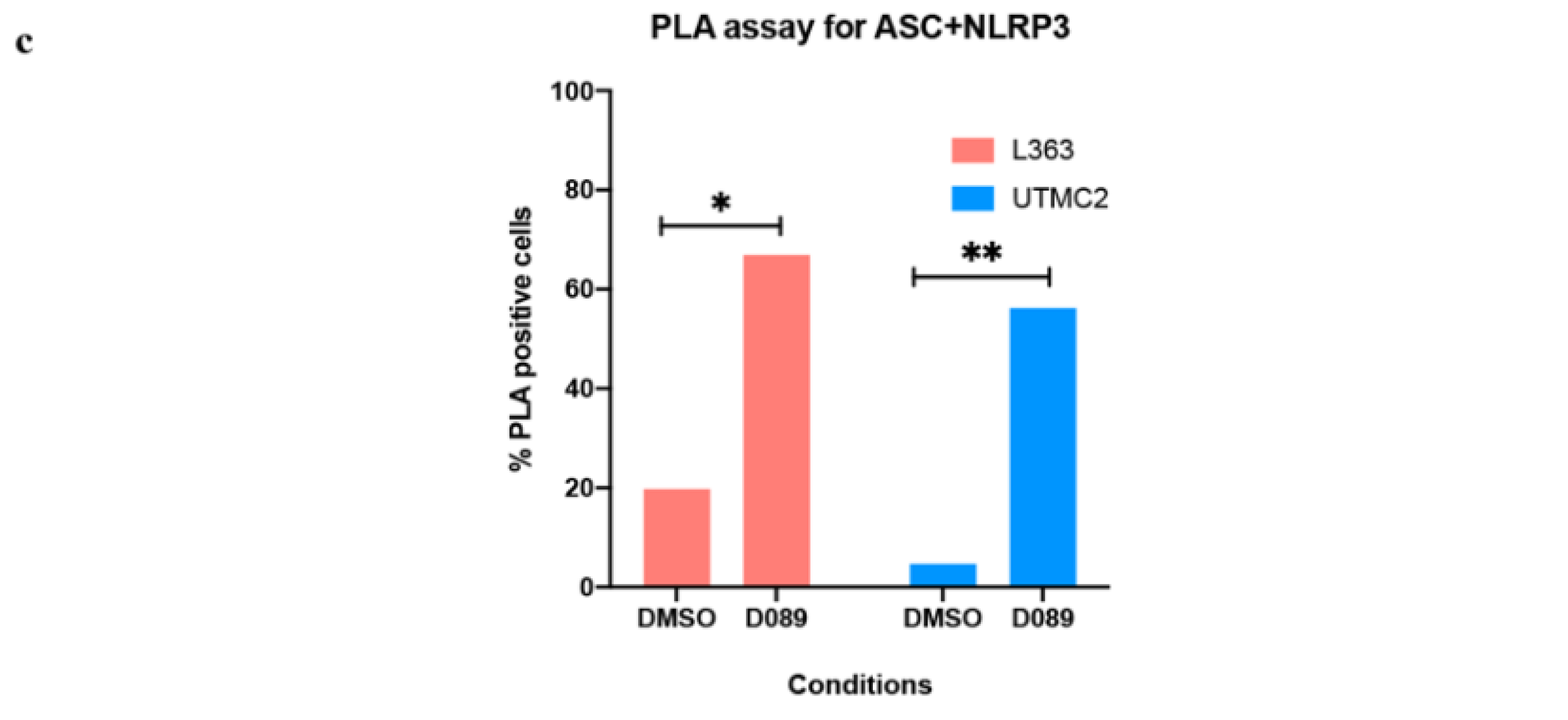
2.5. D089 Triggered Pyroptosis by Priming the Inflammasome Signaling Pathway
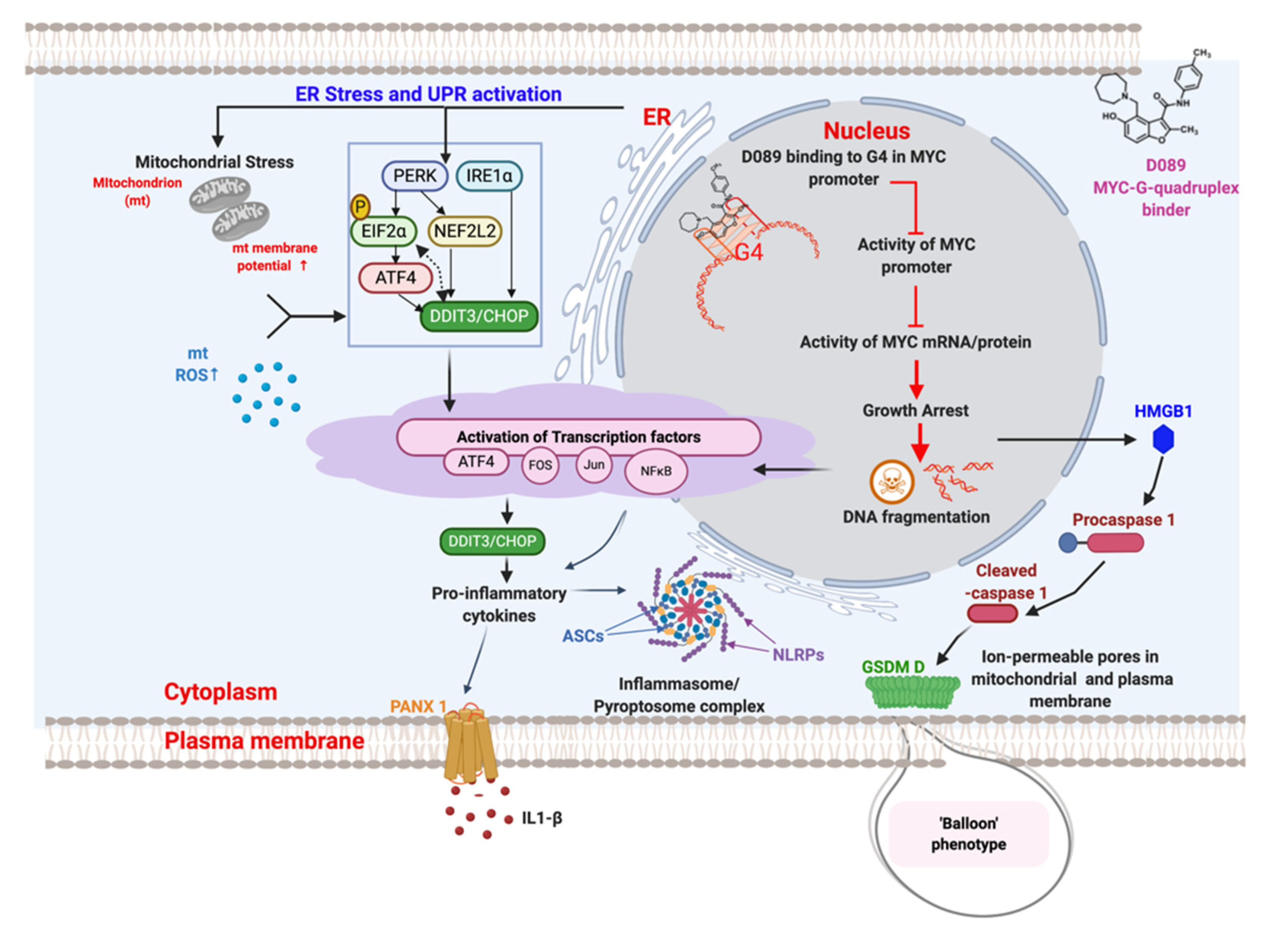
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Culture Conditions
4.2. Drug Compounds
4.3. Plasmid Transfections
4.4. RNA Interference
4.5. Cell Proliferation (MTS) Assay
4.6. Total RNA Isolation and Quantitative Real Time RT-PCR
4.7. RNA Isolation for NanoString nCounter® Gene Expression
4.8. Nanostring nCounter® Gene Expression Quantification
4.9. Protein Extraction and Western Blotting
ASC Oligomerization
4.10. Microscopy Imaging
4.10.1. Immunofluorescence
4.10.2. IncuCyte-Live Cell Imaging
4.11. Proximity Ligation Assay (PLA)
4.12. Flow Cytometry
4.13. LDH Assay
4.14. Quantitative ELISA for Human Interleukin-1 Beta (hu IL-1β)
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Bretones, G.; Delgado, M.D.; Leon, J. Myc and cell cycle control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Dib, A.; Gabrea, A.; Glebov, O.K.; Bergsagel, P.L.; Kuehl, W.M. Characterization of MYC translocations in multiple myeloma cell lines. J. Natl. Cancer Inst. Monogr. 2008, 25–31. [Google Scholar] [CrossRef]
- Holien, T.; Vatsveen, T.K.; Hella, H.; Waage, A.; Sundan, A. Addiction to c-MYC in multiple myeloma. Blood 2012, 120, 2450–2453. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting MYC in multiple myeloma. Leukemia 2018, 32, 1295–1306. [Google Scholar] [CrossRef]
- Simmons, J.K.; Michalowski, A.M.; Gamache, B.J.; DuBois, W.; Patel, J.; Zhang, K.; Gary, J.; Zhang, S.; Gaikwad, S.; Connors, D.; et al. Cooperative Targets of Combined mTOR/HDAC Inhibition Promote MYC Degradation. Mol. Cancer 2017, 16, 2008–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen-Petersen, B.L.; Sears, R.C. Mission Possible: Advances in MYC Therapeutic Targeting in Cancer. BioDrugs 2019, 33, 539–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonsson, T.; Pecinka, P.; Kubista, M. DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res. 1998, 26, 1167–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, D.R.; Chen, X.; Leon, E.C.; Gaikwad, S.M.; Phyo, Z.; Hewitt, W.M.; Alden, S.; Hilimire, T.A.; He, F.; Michalowski, A.M.; et al. Chemical and structural studies provide a mechanistic basis for recognition of the MYC G-quadruplex. Nat. Commun. 2018, 9, 4229. [Google Scholar] [CrossRef] [Green Version]
- Brooks, T.A.; Hurley, L.H. Targeting MYC Expression through G-Quadruplexes. Genes Cancer 2010, 1, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Sedoris, K.C.; Thomas, S.D.; Clarkson, C.R.; Muench, D.; Islam, A.; Singh, R.; Miller, D.M. Genomic c-Myc quadruplex DNA selectively kills leukemia. Mol. Cancer 2012, 11, 66–76. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, K.M.; Saunders, L.B.; Simmons, J.K.; Leon, E.; Calabrese, D.R.; Zhang, S.; Michalowski, A.; Gareiss, P.; Mock, B.A.; Schneekloth, J.S., Jr. Small Molecule Microarrays Enable the Identification of a Selective, Quadruplex-Binding Inhibitor of MYC Expression. ACS Chem. Biol. 2016, 11, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Dickerhoff, J.; Onel, B.; Chen, L.Y.; Chen, Y.W.; Yang, D.Z. Solution Structure of a MYC Promoter G-Quadruplex with 1:6:1 Loop Length. ACS Omega 2019, 4, 2533–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Xing, Z.; Tran, E.J.; Yang, D. DDX5 helicase resolves G-quadruplex and is involved in MYC gene transcriptional activation. Proc. Natl. Acad. Sci. USA 2019, 116, 20453–20461. [Google Scholar] [CrossRef] [Green Version]
- Neidle, S. Human telomeric G-quadruplex: The current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J. 2010, 277, 1118–1125. [Google Scholar] [CrossRef]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Brennan, M.A.; Cookson, B.T. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol. Microbiol. 2000, 38, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef] [PubMed]
- De Gassart, A.; Martinon, F. Pyroptosis: Caspase-11 Unlocks the Gates of Death. Immunity 2015, 43, 835–837. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Vigano, E.; Diamond, C.E.; Spreafico, R.; Balachander, A.; Sobota, R.M.; Mortellaro, A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat. Commun. 2015, 6, 8761. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M.; Kanneganti, T.D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 2016, 16, 7–21. [Google Scholar] [CrossRef]
- Tupik, J.D.; Nagai-Singer, M.A.; Allen, I.C. To protect or adversely affect? The dichotomous role of the NLRP1 inflammasome in human disease. Mol. Asp. Med. 2020, 100858. [Google Scholar] [CrossRef]
- Vanaja, S.K.; Rathinam, V.A.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Jiang, S.; Zhang, Y.; Li, P.; Wang, K. The Multifaceted Roles of Pyroptotic Cell Death Pathways in Cancer. Cancers 2019, 11, 1313. [Google Scholar] [CrossRef] [Green Version]
- Wiggins, K.A.; Parry, A.J.; Cassidy, L.D.; Humphry, M.; Webster, S.J.; Goodall, J.C.; Narita, M.; Clarke, M.C.H. IL-1alpha cleavage by inflammatory caspases of the noncanonical inflammasome controls the senescence-associated secretory phenotype. Aging Cell 2019, 18, e12946. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Li, X.; Liu, Y.; Xia, Y.; Chang, R.; Zhang, C. Inflammasome inhibitors: Promising therapeutic approaches against cancer. J. Hematol. Oncol. 2019, 12, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.Z.; Jiang, J.X.; Gao, Y.; Shi, T.S.; Zhu, X.B.; Zhang, K.J.; Lu, K.; Xue, B. Research progress of the relationship between pyroptosis and disease. Am. J. Transl. Res. 2018, 10, 2213–2219. [Google Scholar] [PubMed]
- Nagarajan, K.; Soundarapandian, K.; Thorne, R.F.; Li, D.; Li, D. Activation of Pyroptotic Cell Death Pathways in Cancer: An Alternative Therapeutic Approach. Transl. Oncol. 2019, 12, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Pyroptosis a cell death modality of its kind? Eur. J. Immunol. 2010, 40, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Pelegrin, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef]
- Cheng, S.B.; Nakashima, A.; Huber, W.J.; Davis, S.; Banerjee, S.; Huang, Z.; Saito, S.; Sadovsky, Y.; Sharma, S. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis. 2019, 10, 927. [Google Scholar] [CrossRef]
- Boddupally, P.V.; Hahn, S.; Beman, C.; De, B.; Brooks, T.A.; Gokhale, V.; Hurley, L.H. Anticancer activity and cellular repression of c-MYC by the G-quadruplex-stabilizing 11-piperazinylquindoline is not dependent on direct targeting of the G-quadruplex in the c-MYC promoter. J. Med. Chem. 2012, 55, 6076–6086. [Google Scholar] [CrossRef] [Green Version]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Cnop, M.; Ladriere, L.; Hekerman, P.; Ortis, F.; Cardozo, A.K.; Dogusan, Z.; Flamez, D.; Boyce, M.; Yuan, J.; Eizirik, D.L. Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic beta-cell dysfunction and apoptosis. J. Biol. Chem. 2007, 282, 3989–3997. [Google Scholar] [CrossRef] [Green Version]
- Fink, S.L.; Cookson, B.T. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol. 2007, 9, 2562–2570. [Google Scholar] [CrossRef]
- Feltham, R.; Vince, J.E.; Lawlor, K.E. Caspase-8: Not so silently deadly. Clin. Transl. Immunol. 2017, 6, e124. [Google Scholar] [CrossRef]
- Jorgensen, I.; Zhang, Y.; Krantz, B.A.; Miao, E.A. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 2016, 213, 2113–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Zha, J.; Wang, Y.; Liu, W.; Yang, X.; Yu, P. Tissue damage-associated “danger signals” influence T-cell responses that promote the progression of preneoplasia to cancer. Cancer Res. 2013, 73, 629–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizato, N.; Luzete, B.C.; Kiffer, L.F.M.V.; Correa, L.H.; Santos, I.D.; Assumpcao, J.A.F.; Ito, M.K.; Magalhaes, K.G. Omega-3 docosahexaenoic acid induces pyroptosis cell death in triple-negative breast cancer cells. Sci. Rep. 2018, 8, 1952. [Google Scholar] [CrossRef]
- Huang, Z.; Zhong, Z.; Zhang, L.; Wang, X.; Xu, R.; Zhu, L.; Wang, Z.; Hu, S.; Zhao, X. Down-regulation of HMGB1 expression by shRNA constructs inhibits the bioactivity of urothelial carcinoma cell lines via the NF-kappaB pathway. Sci. Rep. 2015, 5, 12807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, Z.; Hu, B.; Wu, L.; Jin, S.; Ma, X.; Zhang, J.; Wang, A. Unfractionated Heparin Alleviates Human Lung Endothelial Barrier Dysfunction Induced by High Mobility Group Box 1 Through Regulation of P38-GSK3beta-Snail Signaling Pathway. Cell Physiol. Biochem. 2018, 46, 1907–1918. [Google Scholar] [CrossRef]
- Liu, Z.; Gan, L.; Xu, Y.; Luo, D.; Ren, Q.; Wu, S.; Sun, C. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-kappaB/GSDMD signal in mice adipose tissue. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- He, W.-t.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Yang, D.; He, Y.; Munoz-Planillo, R.; Liu, Q.; Nunez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef] [Green Version]
- Crespo Yanguas, S.; Willebrords, J.; Johnstone, S.R.; Maes, M.; Decrock, E.; De Bock, M.; Leybaert, L.; Cogliati, B.; Vinken, M. Pannexin1 as mediator of inflammation and cell death. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.V.; Danford, F.L.; Gokhale, V.; Hurley, L.H.; Brooks, T.A. Demonstration that drug-targeted down-regulation of MYC in non-Hodgkins lymphoma is directly mediated through the promoter G-quadruplex. J. Biol. Chem. 2011, 286, 41018–41027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, D.; Saha, P.; Das, T.; Dash, J. Target guided synthesis using DNA nano-templates for selectively assembling a G-quadruplex binding c-MYC inhibitor. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Hu, M.H.; Wang, Y.Q.; Yu, Z.Y.; Hu, L.N.; Ou, T.M.; Chen, S.B.; Huang, Z.S.; Tan, J.H. Discovery of a New Four-Leaf Clover-Like Ligand as a Potent c-MYC Transcription Inhibitor Specifically Targeting the Promoter G-Quadruplex. J. Med. Chem. 2018, 61, 2447–2459. [Google Scholar] [CrossRef]
- Liu, R.; Wang, Y.; Li, B.; Wang, H.; Guan, F.; Tan, Z.; Li, X. Screening differentially expressed proteins from co-cultured hematopoietic cells and bone marrow-derived stromal cells by quantitative proteomics (SILAC) method. Clin. Proteom. 2019, 16, 32. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.M.; Galson, D.L.; Roodman, G.D.; Ouyang, H. Resveratrol triggers the pro-apoptotic endoplasmic reticulum stress response and represses pro-survival XBP1 signaling in human multiple myeloma cells. Exp. Hematol. 2011, 39, 999–1006. [Google Scholar] [CrossRef] [Green Version]
- Nikesitch, N.; Lee, J.M.; Ling, S.; Roberts, T.L. Endoplasmic reticulum stress in the development of multiple myeloma and drug resistance. Clin. Transl. Immunol. 2018, 7, e1007. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Guo, X.; Ge, Q.; Zhao, Y.; Mu, H.; Zhang, J. ER Stress Activates the NLRP3 Inflammasome: A Novel Mechanism of Atherosclerosis. Oxid. Med. Cell Longev. 2019, 2019, 3462530. [Google Scholar] [CrossRef]
- Yanagisawa, S.; Katoh, H.; Imai, T.; Nomura, S.; Watanabe, M. The relationship between inflammasomes and the endoplasmic reticulum stress response in the injured spinal cord. Neurosci. Lett. 2019, 705, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Abdul Aziz, A.; Bowles, K.; Rushworth, S. High NRF2 expression controls endoplasmic reticulum stress induced apoptosis in multiple myeloma. Cancer Lett. 2018, 412, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollberger, G.; Strittmatter, G.E.; Garstkiewicz, M.; Sand, J.; Beer, H.D. Caspase-1: The inflammasome and beyond. Innate Immun. 2014, 20, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.P.; Gao, W.Q.; Shi, X.Y.; Ding, J.J.; Liu, W.; He, H.B.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Tripathi, A.; Shrinet, K.; Kumar, A. HMGB1 protein as a novel target for cancer. Toxicol. Rep. 2019, 6, 253–261. [Google Scholar] [CrossRef]
- Guo, X.; He, D.; Zhang, E.; Chen, J.; Chen, Q.; Li, Y.; Yang, L.; Yang, Y.; Zhao, Y.; Wang, G.; et al. HMGB1 knockdown increases MM cell vulnerability by regulating autophagy and DNA damage repair. J. Exp. Clin. Cancer Res. 2018, 37, 205. [Google Scholar] [CrossRef] [Green Version]
- Simmons, J.K.; Patel, J.; Michalowski, A.; Zhang, S.; Wei, B.R.; Sullivan, P.; Gamache, B.; Felsenstein, K.; Kuehl, W.M.; Simpson, R.M.; et al. TORC1 and class I HDAC inhibitors synergize to suppress mature B cell neoplasms. Mol. Oncol. 2014, 8, 261–272. [Google Scholar] [CrossRef]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The role of pyroptosis in cancer: Pro-cancer or pro-“host”? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slomp, A.; Peperzak, V. Role and Regulation of Pro-survival BCL-2 Proteins in Multiple Myeloma. Front. Oncol. 2018, 8, 533. [Google Scholar] [CrossRef] [PubMed]
- Pezuk, J.A. Pyroptosis in combinatorial treatment to improve cancer patients’ outcome, is that what we want? EBioMedicine 2019, 41, 17–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Wang, Y.; Yang, D.; Gong, Y.; Rao, F.; Liu, R.; Danna, Y.; Li, J.; Fan, J.; Chen, J.; et al. A PLK1 kinase inhibitor enhances the chemosensitivity of cisplatin by inducing pyroptosis in oesophageal squamous cell carcinoma. EBioMedicine 2019, 41, 244–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactome Pathways | Raw (p Value) | FDR |
|---|---|---|
| The NLRP1 inflammasome (R-HAS-844455) | 9.04 × 10−5 | 2.30 × 10−3 |
| TFAP2 (AP-2) family regulates transcription of cell cycle factors (R-HSA-8866911) | 1.89 × 10−4 | 4.36 × 10−3 |
| BH3-only proteins associate with and inactivate anti-apoptotic BCL-2 members (R-HSA-111453) | 4.34 × 10−6 | 1.86 × 10−4 |
| SOS-mediated signaling (R-HSA-112412) | 2.52 × 10−4 | 5.51 × 10−3 |
| Activated NTRK3 signals through PI3K (R-HSA-9603381) | 2.52 × 10−4 | 5.46 × 10−3 |
| Activation of the AP-1 family of transcription factors (R-HSA-450341) | 7.49 × 10−6 | 2.93 × 10−4 |
| siRNA | Catalog Number |
|---|---|
| siGENOME Human CASP3 siRNA-SMARTpool | M-004307-02-0010 |
| siGENOME Human CASP1 siRNA-SMARTpool | M-004401-03-0010 |
| siGENOME Non-Targeting siRNA Pool | D-001206-14-50 |
| Primers | Forward | Reverse |
|---|---|---|
| MYC | TGAGGAGACACCGCCCAC | CAACATCGATTTCTTCCTCATCTTC |
| DDIT3 | CAGAACCAGCAGAGGTCACA | AGCTGTGCCACTTTCCTTTC |
| B2M | GCTATCCAGCGTACTCCAAAG | GCTGAAAGACAAGTCTGAATG |
| Antibodies | Company | Catalog Number |
|---|---|---|
| c-Myc | Abcam | ab-32072 |
| p16 for Immunofluorescence | Cell Signaling Technology | 92803 |
| p16 for Immunoblotting | SantaCruz Biotechnologies | sc-1661 |
| p21 for Immunofluorescence | Cell Signaling Technology | 2947 |
| γH2AX for Immunofluorescence | Cell Signaling Technology | 9718 |
| p-EIF2α | Cell Signaling Technology | 3398 |
| EIF2α | Cell Signaling Technology | 9722 |
| PERK | Cell Signaling Technology | 5683 |
| p-PERK | Cell Signaling Technology | 3179 |
| XBP1s | Cell Signaling Technology | 12782 |
| ATF4 | Cell Signaling Technology | 11815 |
| DDIT3/CHOP | Cell Signaling Technology | 2895 |
| IRE1α | Cell Signaling Technology | 3294 |
| NFE2L2/NRF2 | Cell Signaling Technology | 12721 |
| Grp78/Bip/ HSP5A | Cell Signaling Technology | 3177 |
| Caspase 1 | Cell Signaling Technology | 2225 |
| Caspase 3 | Cell Signaling Technology | 9662 |
| Caspase 8 | Cell Signaling Technology | 9746 |
| Caspase 9 | Cell Signaling Technology | 9502 |
| GSDMD | Cell Signaling Technology | 97558 |
| NLRP3 | Cell Signaling Technology | 13158 |
| GAPDH | Cell Signaling Technology | 5174 |
| ASC | SantaCruz Biotechnologies | sc-514414 |
| PANX1 | Cell Signaling Technology | 91137 |
| HMGB1 | Cell Signaling Technology | 6893 |
| NFκB | SantaCruz Biotechnologies | sc-8008 |
| Vincullin | Abcam | ab18058 |
| β-actin | Cell Signaling Technology | 3700 |
| Mouse IgG (H+L) | Invitrogen, ThermoFisher Scientific | 62-6520 |
| Rabbit IgG (H+L) | Invitrogen, ThermoFisher Scientific | 31460 |
| Alexa Fluor 488 (anti-mouse) | Abcam | ab150113 |
| Alexa Fluor 488 (anti-rabbit) | Abcam | ab150077 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaikwad, S.M.; Phyo, Z.; Arteaga, A.Q.; Gorjifard, S.; Calabrese, D.R.; Connors, D.; Huang, J.; Michalowski, A.M.; Zhang, S.; Liu, Z.-G.; et al. A Small Molecule Stabilizer of the MYC G4-Quadruplex Induces Endoplasmic Reticulum Stress, Senescence and Pyroptosis in Multiple Myeloma. Cancers 2020, 12, 2952. https://doi.org/10.3390/cancers12102952
Gaikwad SM, Phyo Z, Arteaga AQ, Gorjifard S, Calabrese DR, Connors D, Huang J, Michalowski AM, Zhang S, Liu Z-G, et al. A Small Molecule Stabilizer of the MYC G4-Quadruplex Induces Endoplasmic Reticulum Stress, Senescence and Pyroptosis in Multiple Myeloma. Cancers. 2020; 12(10):2952. https://doi.org/10.3390/cancers12102952
Chicago/Turabian StyleGaikwad, Snehal M., Zaw Phyo, Anaisa Quintanilla Arteaga, Sayeh Gorjifard, David R. Calabrese, Daniel Connors, Jing Huang, Aleksandra M. Michalowski, Shuling Zhang, Zheng-Gang Liu, and et al. 2020. "A Small Molecule Stabilizer of the MYC G4-Quadruplex Induces Endoplasmic Reticulum Stress, Senescence and Pyroptosis in Multiple Myeloma" Cancers 12, no. 10: 2952. https://doi.org/10.3390/cancers12102952