
Synthesis and Characterization of Atmospherically Relevant Hydroxy Hydroperoxides


1
Atmospheric Chemistry Department (ACD), Leibniz Institute for Tropospheric Research (TROPOS), 04315 Leipzig, Germany
2
Eurofins Institut Dr. Appelt Leipzig GmbH, 04317 Leipzig, Germany
*
Author to whom correspondence should be addressed.
Atmosphere 2022, 13(4), 507; https://doi.org/10.3390/atmos13040507
Submission received: 13 January 2022
/
Revised: 5 March 2022
/
Accepted: 14 March 2022
/
Published: 22 March 2022
(This article belongs to the Special Issue The Critical Role of Synthetic Chemistry in Elucidating Mechanisms, Product Identification, and Quantitation in Atmospheric Gas-Phase and Multiphase Chemistry of Volatile Organic Emissions)
Abstract
:Hydroxy hydroperoxides are formed upon OH oxidation of volatile organic compounds in the atmosphere and may contribute to secondary organic aerosol growth and aqueous phase chemistry after phase transfer to particles. Although the detection methods for oxidized volatile organic compounds improved much over the past decades, the limited availability of synthetic standards for atmospherically relevant hydroxy hydroperoxides prevented comprehensive investigations for the most part. Here, we present a straightforward improved synthetic access to isoprene-derived hydroxy hydroperoxides, i.e., 1,2-ISOPOOH and 4,3-ISOPOOH. Furthermore, we present the first successful synthesis of an α-pinene derived hydroxy hydroperoxide. All products were identified by 1H, 13C NMR spectroscopy for structure elucidation, additional 2D NMR experiments were performed. Furthermore, gas-phase FTIR- and UV/VIS spectra are presented for the first time. Using the measured absorption cross section, the atmospheric photolysis rate of up to 2.1 × 10−3 s−1 was calculated for 1,2-ISOPOOH. Moreover, we present the investigation of synthesized hydroxy hydroperoxides in an aerosol chamber study by online MS techniques, namely PTR-ToFMS and (NO3−)-CI-APi-ToFMS. Fragmentation patterns recorded during these investigations are presented as well. For the (NO3−)-CI-APi-ToFMS, a calibration factor for 1,2-ISOPOOH was calculated as 4.44 × 10−5 ncps·ppbv−1 and a LOD (3σ, 1 min average) = 0.70 ppbv.
1. Introduction
Secondary organic aerosols (SOA) can constitute a major fraction of atmospheric aerosol mass with implications for climate, visibility and human health [1]. The precursor compounds for this mass fraction are oxidation products of hydrocarbon emissions, dominated by biogenic sources. Among these, methane, isoprene and monoterpenes have the highest source strengths [2], with α-pinene being the most detected and most investigated compound of the latter.
The most abundant oxidant in Earth’s atmosphere is the OH radical [3], which can be produced by the photodissociation of ozone to an excited state oxygen atom O1D and subsequent reaction with water vapor [4]. Another important source is the secondary formation from degradation pathways of oxidized volatile organic compounds, which can lead to radical recycling [5]. OH radicals react rapidly with unsaturated hydrocarbons such as isoprene and monoterpenes. The attack of a double bond or allylic system is commonly preferred over methyl hydrogen abstraction due to a much faster reaction rate. The addition primarily takes place at less substituted positions since the resulting alkyl or allylic radical is more stable [6]. Subsequent addition of oxygen leads to peroxyl radicals. Especially for allylic radicals, the bond dissociation energy of the R-OO bond is comparably small, and the addition of O2 is a reversible process [7,8]. Therefore, in these cases, the constituency of the corresponding alkyl radical widely determines the branching ratio of possible oxidation products [9].
RO2 radicals can undergo both intra- and unimolecular as well as bimolecular reactions in the atmosphere, depending on the oxidation regime as well as the atmospheric lifetime of the RO2 in question. Intramolecular H-shift reactions lead to the formation of hydroperoxide moieties and shift the radical position within the molecule [10,11]. This can drive further oxidation of the carbon backbone and leads to highly oxidized molecules (HOMs). Such intramolecular reactions are reported to occur comparably fast depending on the specific molecular structure [12].
Under low NO conditions and with low overall concentrations of other oxidants, this process might determine the fate of atmospheric RO2 radicals. In regions with higher volatile organic carbon (VOC) concentrations and still under low NO, however, higher concentrations of peroxyl radicals can occur, and bimolecular self- and cross reactions can become a more important process [13,14,15]. Here, two peroxyl radicals can react to yield hydroxy- and carbonyl groups on either of the molecules. For a number of different systems, the formation of RO2 accretion products (ROOR’) has been observed [16,17,18]. In regions influenced by anthropogenic activities, high NO concentrations can result in the fast transfer of an oxygen atom from RO2, leading to the formation of alkoxy radicals that rapidly decompose, resulting in a C-C bond cleavage. In contrast, in rural areas, the abstraction of a H-atom from HO2 radicals can be the major process for RO2 removal as HO2 concentrations can exceed 10 pptv [19]. This reaction terminates the radical chain reaction and results in hydroxy hydroperoxides.
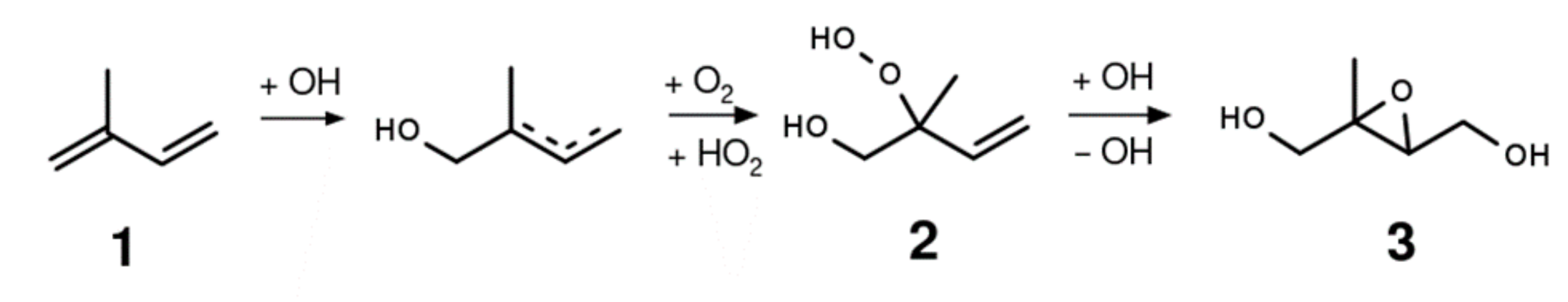
In the case of isoprene (1) oxidation, intermediate products of this class, such as 1-hydroxy-2-hydroperoxy-2-methyl-but-3-ene (1,2-ISOPOOH 2), were already hypothesized 30 years ago [20]. The first reported measurements utilizing proton transfer reaction mass spectrometry (PTR-MS) [21,22], however, are widely ascribed to other isobaric compounds named isoprene epoxy diols (IEPOX 3) as shown in Scheme 1 [23]. Still, isoprene-derived hydroxy hydroperoxides (ISOPOOH) were reportedly measured nearly a decade later with even milder analytical techniques, namely CF3O− chemical ionization atmospheric pressure interface time-of-flight mass spectrometry (CI-APi-ToFMS) [24]. This emphasizes the importance of reliable analytical data based on synthetic standards to confirm reaction pathways and molecular structures.
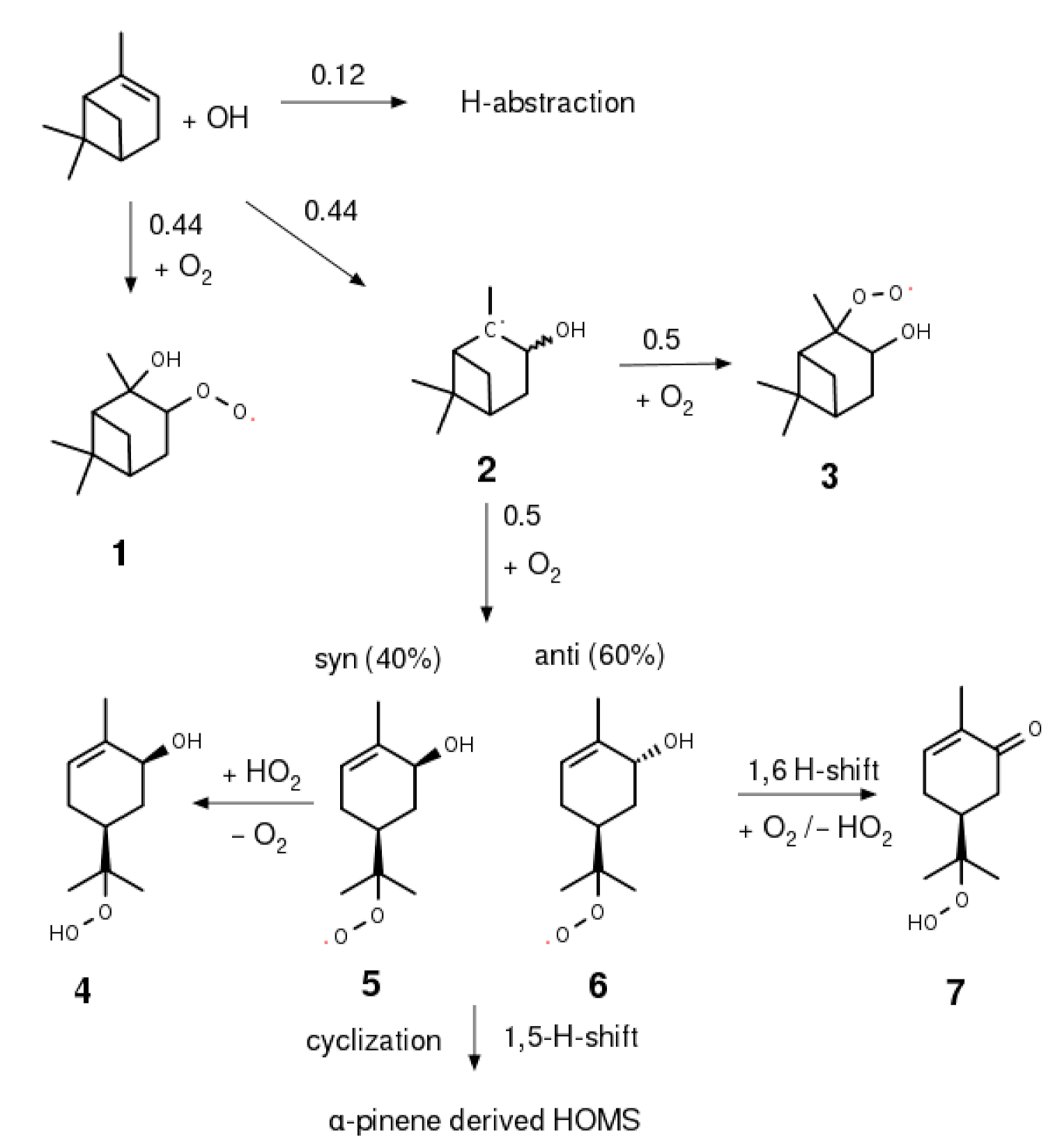
For α-pinene oxidation, the first-generation hydroxy hydroperoxide has not yet been observed. Here, the OH attack can produce two diastereomeric pairs of hydroxy alkyl radicals, depending on the position of the initial OH attack on the double bond, as shown in Scheme 2. The (2-OH, 3-R) isomer directly forms vicinal hydroxy peroxyl groups as in 4, that can undergo bimolecular reactions [25] and consequently, also form hydroxy hydroperoxides. If the radical position is at the tertiary carbon as for the alkyl radical 5, however, a prompt ring opening driven by the ring strain of the four-membered ring can occur [26,27]. This process is estimated to remove about half of the alkyl radicals 5 to form hydroxy peroxyl radicals 8 and 9 upon oxygen addition [28]. The remaining fraction supposedly forms α-hydroxy-peroxyl radicals 6 after thermalization and subsequent oxygen addition.
Vereecken et al. [27] investigated the atmospheric fate of the peroxyl radical resulting from ring opening and estimated small isomerization barriers for 1,6-H-shifts both for syn and anti products. The calculated values for atmospheric rate constants, however, diverge drastically between the respective isomers. For the anti product 9, they result in fast reaction rate constants of up to 2000 s−1 forming further oxidized molecules [29] and eventually leading to the formation of closed-shell products such as carbonyl compound 10. Such high rate constants practically exclude competing bimolecular reaction pathways under atmospheric conditions. The estimated rate constant for the syn conformer 8 was found to be 2.6 s−1 and hence is three orders of magnitude lower. Experimental results from Xu et al. [30,31] support this estimation. By gradually increasing the budget of bimolecular reaction partners as calculated using the master chemical mechanism (MCM), these authors determined 4 ± 2 s−1 as a rate constant for the unimolecular reaction step involved. Nevertheless, a series of different isomers of α-pinene-derived hydroxy hydroperoxides can be hypothesized. Their formation could occur after ring opening as in compound 7 or without ring opening, corresponding to the peroxyl radicals 4 and 6. The lack of authentic standards is a major limitation and leaves uncertainties about these reaction pathways and the atmospheric significance of the closed-shell products.
2. Experimental Methods
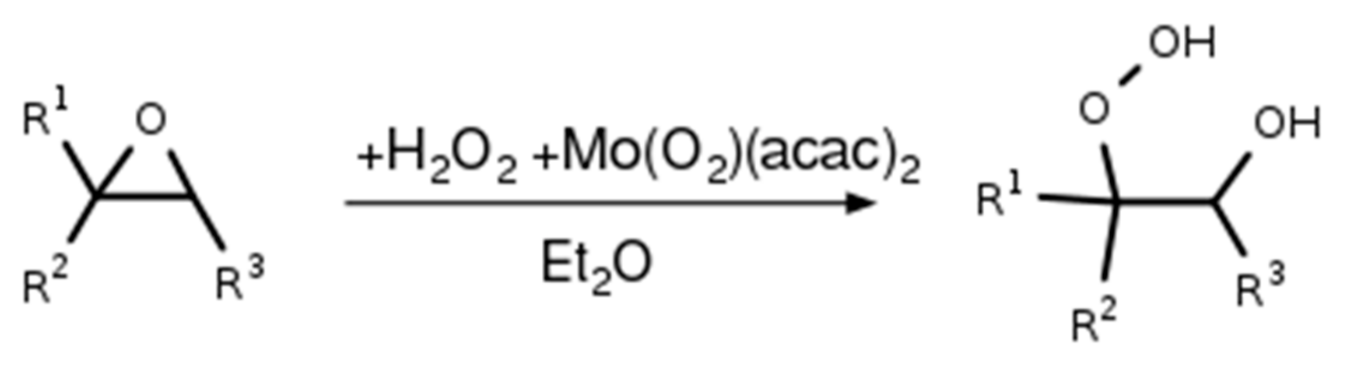
All hydroxy hydroperoxides were synthesized by catalytic ring opening of corresponding epoxides with hydrogen peroxide as shown in Scheme 3.
To avoid hydrolysis in the presence of the catalyst, a water-free solution of hydrogen peroxide in diethyl ether was first prepared as follows. To 15 mL of a 30% hydrogen peroxide solution, sodium chloride was added in excess. The mixture was stirred for 10 min before hydrogen peroxide was extracted with 3 × 20 mL diethyl ether. The organic phases were combined and dried over magnesium sulfate. The concentration of hydrogen peroxide was monitored by titration with acidified potassium permanganate solution and found to be around 1 mol/L.
Nuclear magnetic resonance (NMR) spectra were recorded with a 600 MHz NMR spectrometer (AVANCE III, BRUKER), equipped with a BBO broad-band probe head in deuterated chloroform solution at room temperature. To measure gas-phase IR and UV/VIS spectra, the analytical standards were placed in a small glass flask and constantly flushed with 1000 cm3 min−1 nitrogen. The gas flow was diluted directly after leaving the flask to avoid high residence times and, thus, possible surface reactions. The gas stream was flushed continuously through the measurement cell of an FT-IR spectrometer (Thermo, Nicolet 6700), equipped with a White mirror system (20 m path length). A total of 1000 scans was recorded in the range of 3500–750 cm−1 with a resolution of 1 cm−1 using a mercury telluride detector. Similarly, the air stream was flushed through a UV cell (volume: 5 L, path length 4.22 m) and for each synthetic standard 3 spectra in the range of 200–800 nm were recorded with a resolution of 0.1 nm.
The photolysis rates of methyl hydroperoxide and 1,2-ISOPOOH are calculated offline using the measured absorption cross sections and assumed quantum yields of unity for both hydroperoxides. To calculate the photolysis rate, the Tropospheric Ultraviolet and Visible (TUV) radiation model version 5.3.2 [32] was used. The solar zenith angle was calculated for 21 March at 0° latitude, 500 m height. To retrieve a parametrization to implement in MCM, an additional run at 45° latitude at an altitude of 500 m for 1 July was performed [33].
2.1. Synthesis of the Isoprene-Derived β-Hydroxy Hydroperoxides
All the synthesis of the isoprene-derived β-hydroxy hydroperoxides performed in the present work can be depicted as shown in Scheme 3.
2.2. Synthesis of 1,2-ISOPOOH
In a 50 mL flask 20 mL of the 0.89 mol/L etheric hydrogen peroxide (17.9 mmol; 1.5 equiv) solution was added to 1.00 g (11.9 mmol) 1-epoxy-2-methyl-but-1-ene. After addition of 20 mg (0.06 mmol; 0.005 equiv) bis(acetylacetonato)dioxomolybdenum(VI) the reaction mixture was stirred for 1 h and monitored with thin layer chromatography (TLC). Under reduced pressure, the diethyl ether was removed until about 1.5 mL remained in the flask. The residue was immediately purified by flash column chromatography (ethyl acetate, n-hexane (1:1)) to give 0.72 g of a colorless viscous liquid (yield: 51%). RF,TLC = 0.38 (ethyl acetate: n-hexane, 2:1 (v:v)). 1H-NMR (600 MHz, CDCl3) δ: 5.91 (dd, J = 18, 11.4 Hz, 1H), 5.26 (dd, J = 18, 1.2 Hz, 1H), 5.23 (dd, J = 11.4, 1.2 Hz, 1H), 3.61 (d, J = 12 Hz, 1H), 3.75 (d, J = 12 Hz, 1H), 1.26 (s, 3H).
2.3. Synthesis of 4,3-ISOPOOH
As it is not commercially available, 3-epoxy-2-methyl-but-1-ene was synthesized from methacrolein via a Johnson–Corey–Chaykovsky reaction [34,35]. Briefly, in a 100 mL flask, 8.85 g (220 mmol, 7.5 equiv) sodium hydroxide was added to a solution of 5.55 g (29.5 mmol, 1 equiv) trimethylsulfonium methyl sulfate in 55 mL dichloromethane. The mixture was stirred at room temperature, while a solution of 2.27 g (32.45 mmol, 1.1 equiv) methacrolein in 25 mL dichloromethane was added dropwise. After 3.5 h, the reaction mixture was cooled to 273 K, and 20 mL water was added. The aqueous phase was removed, and the remaining organic phase was washed with portions of 15 mL of water until the residual aqueous phase showed neutral pH. Subsequently, the solvent was removed under reduced pressure to give 2.1 g (yield = 85%) of a colorless liquid. RF,TLC = 0.46 (ethyl acetate: n-hexane, 2:1 (v:v)).
The transformation to 4-hydroxy-3-hydroperoxy-2-methyl-but-1-ene was carried out similar to 1,2-ISOPOOH. 1.0 g (11.9 mmol) 3-epoxy-2-methyl-but-1-ene was added to 17 mL of a 0.85 mol/L solution of hydrogen peroxide (14.3 mmol, 1.2 equiv) in diethyl ether. The reaction was stirred for 1.5 h after addition of 20 mg (0.06 mmol; 0.005 equiv) bis(acetylacetonato)dioxomolybdenum (VI). Subsequently, the diethyl ether was removed under reduced pressure and the residue was purified immediately by column chromatography (ethyl acetate: n-hexane, 1:1; (v:v)) to give 0.2 g of a colorless viscous liquid (yield 14%). RF,TLC = 0.65 (ethyl acetate: n-hexane, 2:1 (v:v)). 1H-NMR (600 MHz, CDCl3) δ: 5.05 (m, 2H), 4.51 (t, J = 6 Hz, 1H), 3.74 (s, 1H), 3.73 (d, J = 17.4 Hz, 1H), 1.76 (s, 3H).
2.4. Synthesis of α-Pinene Hydroxy Hydroperoxide
In a 50 mL flask 19 mL of the 1 mol/L etherial hydrogen peroxide (15 mmol; 1.5 equiv) solution was added to 1.52 g (10 mmol) α-pinene-oxide. After addition of 16 mg (0.005 mmol; 0.05 equiv) bis(acetylacetonato)dioxomolybdenum (VI) the reaction mixture was stirred for 90 min. Under reduced pressure, the diethyl ether was removed until about 1.5 mL remained in the flask. The residue was immediately purified by column chromatography (ethyl acetate, n-hexane) to give 0.29 g of a colorless viscous liquid (yield: 17%) RF,TLC = 0.46 (ethyl acetate: n-hexane, 2:1 (v:v))). 1H-NMR (600 MHz, CDCl3) δ: 5.56 (d, J = 3 Hz, 1H), 4.04 (s, 1H), 2.17 (m, 1H), 2.02 (s, 1H), 2.00 (s, 1H), 1.76 (s, 3H), 1.69 (m, 1H), 1.33 (m, 1H), 1.23 (s, 3H), 1.04 (s, 3H).
2.5. ACD-C Aerosol-Chamber Setup
The aerosol chamber of the Atmospheric Chemistry Department (ACD) at TROPOS (ACD Chamber, or ACD-C) was used to characterize the analytical standards with online MS-techniques. The 19 m3 chamber made of fluorinated ethylene propylene was flushed with purified air at 200 L·min−1 for at least 18 h prior to each experiment. The chamber was kept at a constant temperature of 293 ± 2 K at 1000 mbar for all experiments. A more detailed description of the chamber can be found in previous publications [36]. The synthetic standards of organic hydroxy hydroperoxides were characterized using NO3−-CI-APi-ToFMS [37]. The resulting spectra were averaged over a one-minute time period to account for low cluster stabilities. Subsequently, a suitable mass calibration was performed automatically for each averaged spectrum. After integration, all signals were normalized by the sum of the first 3 primary ion clusters m/z 62, 125 and 188, representing NO3−, (HNO3)NO3− and (HNO3)2NO3− respectively. Additionally, a proton transfer reaction time-of-flight mass spectrometer (PTR-ToFMS) [38] was used to characterize the analytical standards as well as the decomposition patterns. Specifically, for this study, an inlet temperature of 60 °C and drift pressure of 2.20 mbar were used. Here, ion signals were integrated from a 10 s averaged spectrum and subsequently normalized by the primary ion signal, that was calculated from the most abundant 18O isotope peak. To account for possible nucleation events or any other unwanted complication by particles in the chamber, the particle number concentration was monitored with a TROPOS-style Mobility Particle Size Spectrometer (MPSS) [39]. Nucleation events did not occur according to our measurements.
Syringe injection of pure peroxides or their ether solutions into the chamber resulted in large variations in gas-phase concentrations. Consequently, the samples of analytical standards were introduced into the chamber setup with a gentle gas stream. Briefly, a 50 mL flask containing the viscous organic peroxide was covered with the top of a gas wash bottle, bringing the gas flow as close as possible to the surface of the liquid droplet. A constant airflow of 10 L·min−1 was flushed over the synthetic standard. After a fixed time period, the flask containing ISOPOOH was removed, and an empty flask was added, thus allowing residual ISOPOOH to be flushed into the chamber without further loss to walls and tubing. The mass loss in the flask accounted for any peroxide transferred to the gas phase. The chamber wall loss was determined from a 15 min time period following the last injection. Consequently, all time series were corrected by calculating the wall loss for every time step and adding the total wall loss to the original signal. Calibration factors were calculated by linear regression of the resulting change of the normalized signal after each injection step to the corresponding mixing ratio in the chamber calculated from the mass loss in the flask. To calculate the limit of detection (LOD), a blank experiment with a clean chamber was performed over one hour. The standard deviation (σ) of the normalized signal was calculated, and the limit of detection was determined as three times the standard deviation divided by the calibration factor.
3. Results and Discussion
3.1. Product Characterization following the 1,2-ISOPOOH Synthesis
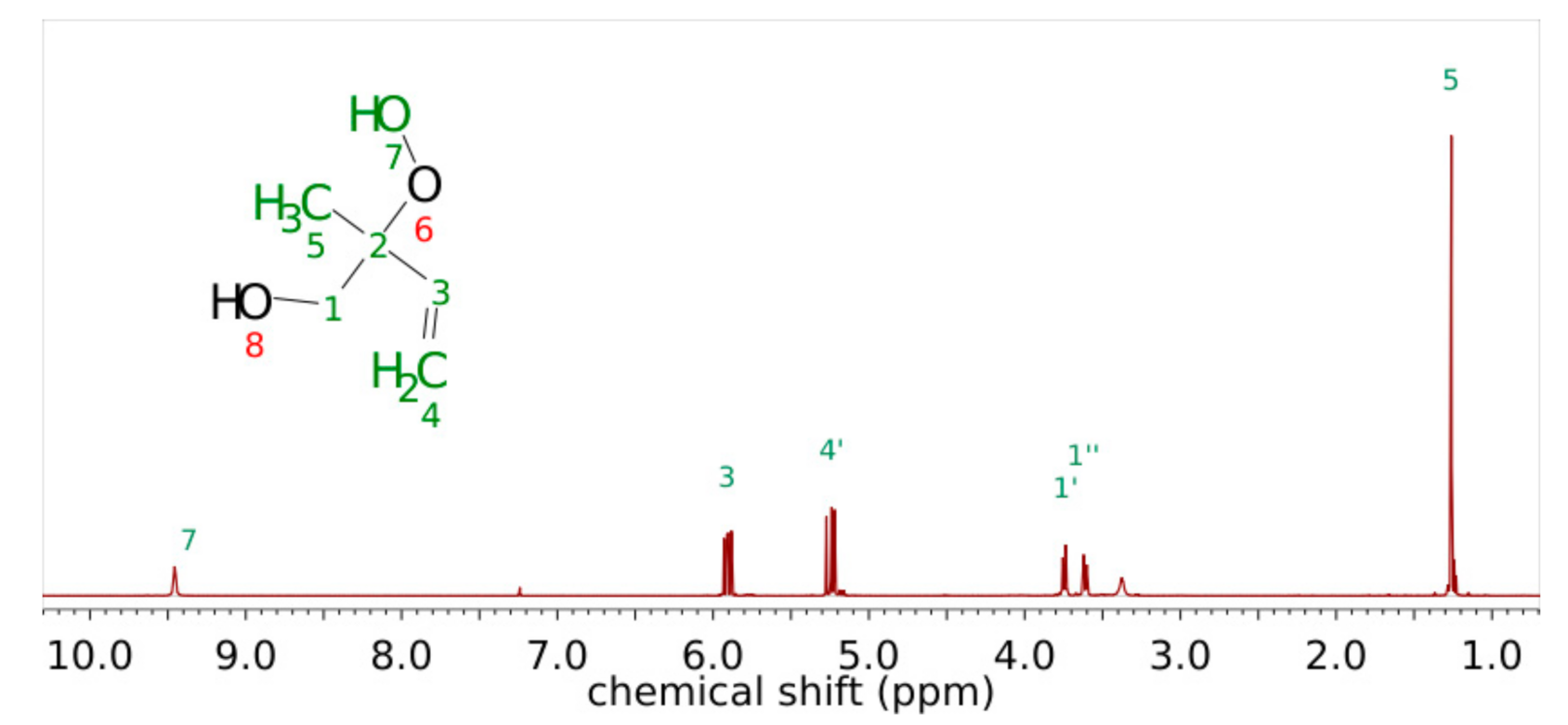
The synthesis of both 1,2-ISOPOOH and 4,3-ISOPOOH have been reported previously. Unfortunately, the currently available reports lack detailed descriptions of the experimental conditions as well as full analytical characterization. Riva et al. [40] reported the successful synthesis using phosphomolybdic acid as a catalyst following the procedure of Li et al. [41], who investigated the applied procedure for a variety of compounds, mostly cyclic aliphatic and olefinic compounds. For α,β unsaturated compounds, they reported lower yields of the hydroxy hydroperoxide together with a complex product spectrum and the reaction mixture turning deep blue during the reaction time. When applying phosphomolybdic acid to the reaction of isoprene epoxide and hydrogen peroxide, the same effect was observed. Presumably, α,β unsaturated hydroxy hydroperoxides are prone to further reaction with the comparatively acidic catalyst phosphomolybdic acid, as has been proposed before [42]. As a consequence, in the present study, the much less acidic [MoO2(acac)2] was used as a catalyst. The reaction mixture turned yellow upon the addition of the catalyst without any further color change. Furthermore, the reaction produced the desired hydroxy hydroperoxides in high yields with fewer byproducts to remove. The product was purified by column chromatography, and the structure was confirmed by NMR. Figure 1 shows the 1H-NMR spectrum, which features proton shifts and integrals consistent with the structure as well as the number and relative integrals of the previously reported spectrum of 1,2-ISOPOOH [43].
The spectrum shows two peaks in the typical olefin region at 5.90 ppm and 5.24 ppm with the integral of one and two protons, respectively. Moreover, the signal for the methyl group is shifted compared to the isoprene NMR spectrum [44], indicating a residual double bond, which is not located in the vicinal position of the methyl group. The signals of the oxidized double bond, in contrast, are shifted to 3.61 and 3.75 ppm, respectively, which is in good agreement with the molecular structure. Recently, Dovrou et al. reported the 1H-NMR spectrum of both ISOPOOH isomers in D2O with similar chemical shifts and integrals of the proton signals [45]. In contrast to spectra recorded in deuterated chloroform, the methylene protons H1 are slightly shifted to 3.5 ppm and show only one signal. Zanca et al. investigated the 1H-NMR spectra of isoprene-derived SOA [46] and reported a large number of signals from 3.4 to 3.9 ppm, which were identified as signals of methyl tetrols as the main constituents. An unresolved background, however, was attributed to structurally similar compounds, namely corresponding peroxides with the same carbon backbone. In contrast to terpene oxidation experiments, the isoprene oxidation did not show significant change over time corresponding to progressive oxidation. The NMR signals of ISOPOOH as a key first-generation oxidation product might contribute to these unresolved background signals, and ongoing oxidation from first- to second-generation oxidation products might escape elucidation. The corresponding diol 1,2-dihydroxy-2-methyl-but-3-ene (1,2-ISOPOH) was identified as the main impurity with the olefin signals shifted to 5.77 ppm and 5.18 ppm, respectively, matching the previously reported data [45,47]. The purity of 1,2-ISOPOOH was calculated from the ratio of corresponding signals and determined as ≥97%. The 13C, COSY, HMBC and HSQC spectra can be found in the supporting information Figures S1–S4. The detailed peak assignments and coupling constants are listed in Table S1. Figure S5 shows the resulting PTR-ToFMS and NO3−-CI-APi-ToFMS spectra of 1,2-ISOPOOH.
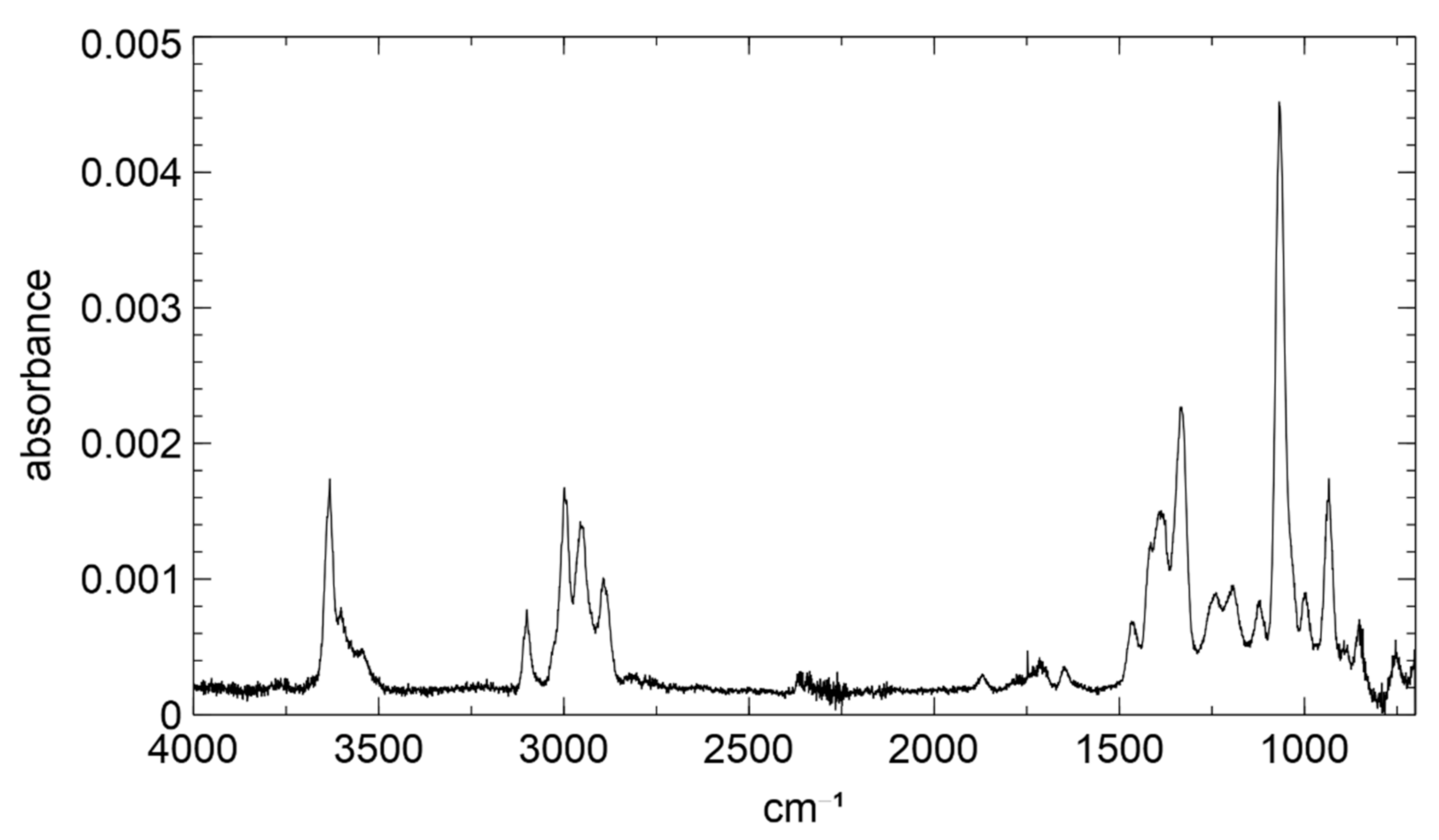
Furthermore, a gas-phase IR-spectrum was recorded and is shown in Figure 2, featuring the characteristic OH-absorption band at 3650 cm−1.
Both the alcoholic and the hydroperoxide O–H bond absorb at similar wavelengths and might equally contribute to this absorption band, as shown by Shreve et al. [48]. The C-H stretch region shows four distinct absorption bands. The two bands at 3100 cm−1 and 3000 cm−1 can also be observed for the isoprene IR spectrum [49] and can be attributed to primary and secondary aliphatic C–H bonds. The bands at 2950 cm−1 and 2890 cm−1, in contrast, show a much-increased absorption in comparison to the isoprene spectrum due to the much stronger absorption of C–H bonds in methylene groups in saturated carbon backbones. A weak absorption was observed in the carbonyl absorption region between 1650 and 1800 cm−1, which might originate from carbonyls formed in the surface reaction of 1,2-ISOPOOH, leading to C4-carbonyls and formaldehyde [50]. Furthermore, the spectrum shows three characteristic absorption bands in the fingerprint region at 935, 1070 and 1330 cm−1, presumably originating from the peroxide moiety. Shreve et al. [48] reported an absorption band at 900 cm−1, which occurred for tert-butanol and disappeared in the spectra of the corresponding hydroperoxide. Instead, an absorption band at 1100 cm−1 occurred for two hydroperoxides, namely cyclohexene hydroperoxide and 1,2,3,4-tetrahydronaphtalene-1-hydroperoxide. In the reported spectra, both absorption bands can be observed, which could be explained by both hydroxy- and hydroperoxide moieties in the structure. In contrast, Vacque et al. [51] reported peroxide absorptions at 845 cm−1 for hydroperoxides. Mohnhaupt et al. reported a similar absorption for peroxide structures at 880 cm−1, which were attributed to coupled C–O and O–O stretching in the C–O–O element [52]. To confirm the origin of these absorption bands, however, additional experiments with isotopically labeled ISOPOOH would be necessary, which is beyond the scope of the present study.
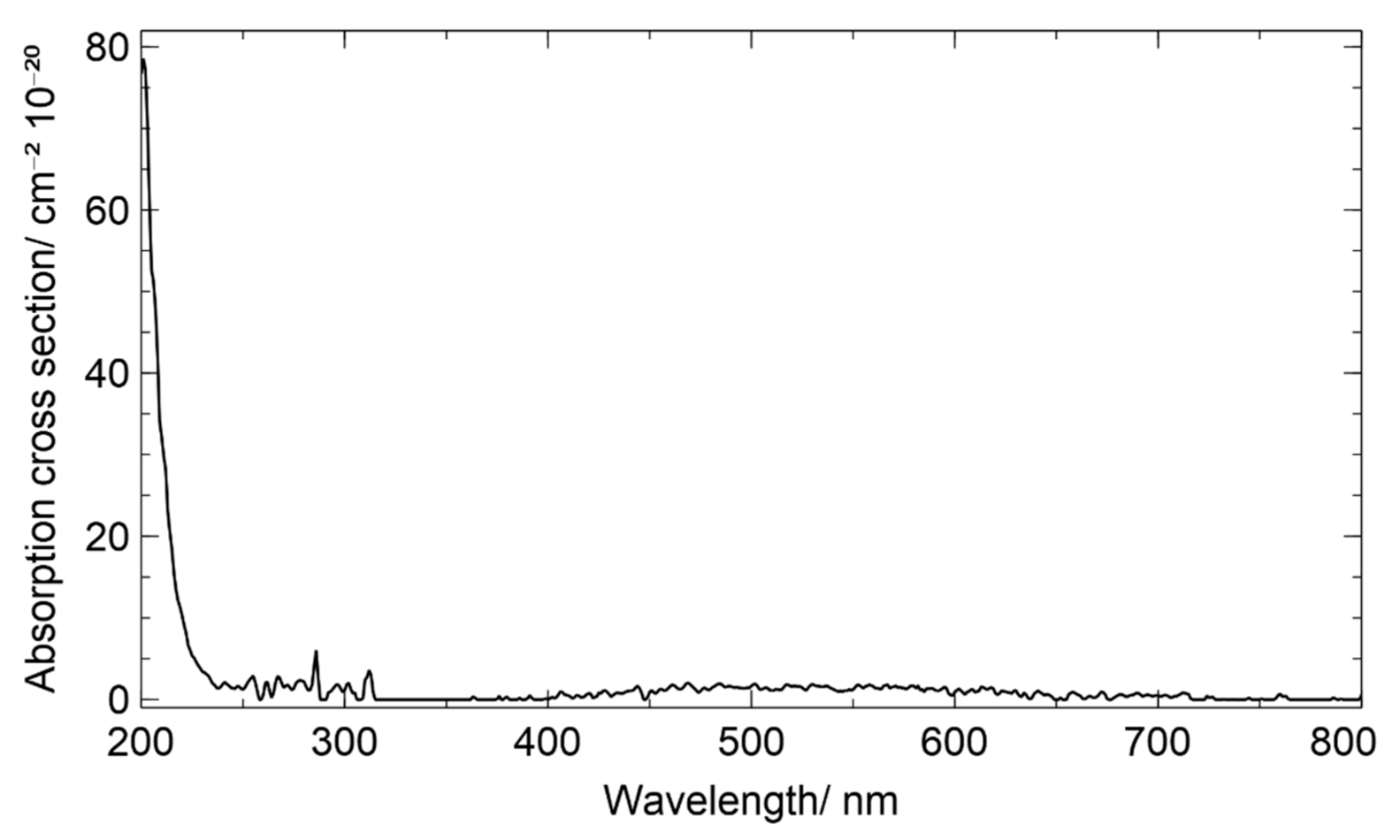
Figure 3 shows the UV/VIS spectrum recorded with a diluted gas stream. Admittedly, exact gas-phase concentration could not be derived due to unknown phase transfer rates between the compartments.
To estimate the gas-phase concentration, an experiment was performed, flushing the analytical standard with the same nitrogen flow over a longer period of time. In 4.5 h of experiment, a total of 7.0 mg of 1,2-ISOPOOH evaporated. Assuming no other reactions take place and a constant phase transfer, a gas-phase concentration of 1.32 × 1014 molecule·cm−3 can be estimated in the gas stream leaving the flask, resulting in the given absorption cross sections with dilution by the carrier gas included.
As 1,2-ISOPOOH does not contain strongly absorbing chromophores in its molecular structure, it is not surprising that the UV/VIS spectrum shows the highest extinction coefficients below 250 nm, similar to other small molecular peroxide compounds. The absorption cross section of 1,2-ISOPOOH at 214 nm was measured to be 27.43 × 10−20 cm2 molecule−1, which is between the reported cross sections of methyl hydroperoxide and hydrogen peroxide, (22.51 ± 0.78) × 10−20 cm2 molecule−1 and (33.04 ± 2.17) × 10−20 cm2 molecule−1, respectively [53]. For atmospheric conditions, however, the comparatively small absorption between 400 and 650 nm with the maximum of 2.03 × 10−20 cm2 molecule−1 at 470 nm might be of significant importance, as sunlight with wavelengths shorter than 290 nm is absorbed efficiently by ozone and does not reach the bottom of the troposphere. For methyl hydroperoxide weaker absorption cross sections than 2 × 10−22 cm2 molecule−1 have been reported for the region between 350 and 650 nm [54]. To the authors’ best knowledge, this is the first quantitative gas-phase ISOPOOH UV/VIS absorption spectrum currently available for the wavelength range 200 to 800 nm.
The atmospheric photolysis of ISOPOOH is usually neglected due to the fast reaction with atmospheric oxidants [55,56]. This assumption, however, has not yet been confirmed with analytical data. The measurement of the absorption cross section is a crucial parameter to retrieve atmospheric lifetimes for both OH oxidation and photolysis. However, the light absorption properties of organic hydroperoxides are widely treated similarly. To input photolysis rates despite missing data, hydroperoxides such as 1,2-ISOPOOH are usually treated in atmospheric models, such as the MCM, using the same parametrization as methyl hydroperoxide (CH3OOH) [33].
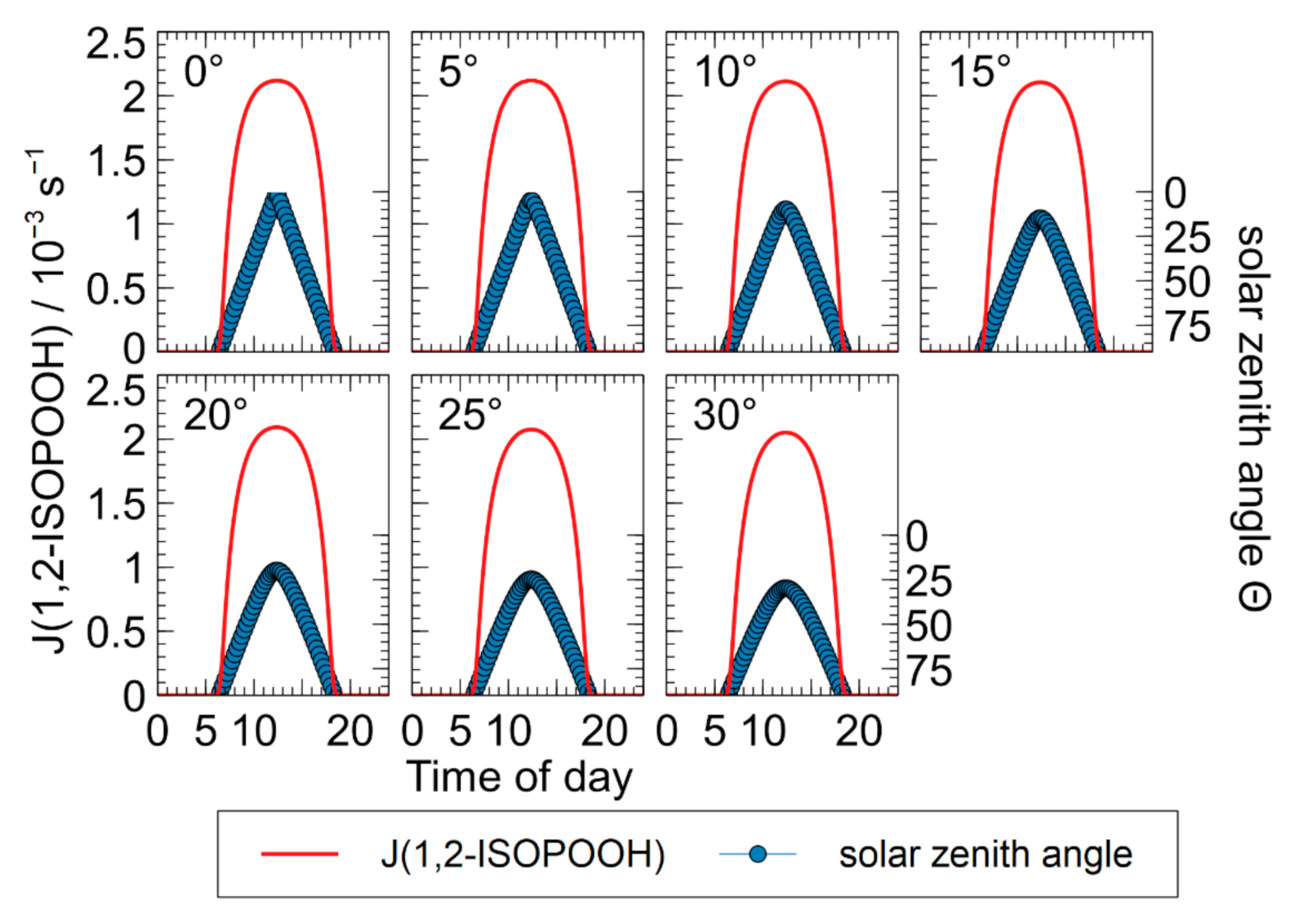
To estimate the atmospheric lifetime of ISOPOOH, its photolysis rate was calculated over the course of a day using the TUV-radiation model. Since the major fraction of isoprene is emitted over the rainforests of the equator region [2], the calculation was performed for 21 March between 0° and 30° latitude under clear sky conditions to evaluate atmospheric photolysis. The calculated maximum photolysis rate at noon ranges between 2.05 × 10−3 s−1 and 2.12 × 10−3 s−1, as shown in Figure 4. For comparison, the photolysis rate of methyl hydroperoxide was calculated for the same case at 0° latitude with the absorption cross section adapted from Vaghjiana and Ravishankara [53]. The photolysis rate shows a similar diurnal cycle as for 1,2-ISOPOOH shown in Figure 4, with a maximum at noon at 7.56 × 10−6 s−1, which is more than two orders of magnitude smaller than for 1,2-ISOPOOH. To evaluate photolysis as a possible atmospheric sink for ISOPOOH, it is necessary to compare it to OH oxidation. The rate constant of 1,2-ISOPOOH with OH radicals has been reported to be (7.5 ± 1.2) × 10−11 cm3 molecule−1 s−1 [56]. With typical OH radical concentrations of 2 × 106 molecule cm−3 [5], a first-order constant of about 1.5 × 10−4 s−1 can be estimated. Accordingly, while the photolysis rate of methyl hydroperoxide is much lower at all times, the photolysis of 1,2-ISOPOOH might be a process competitive with OH oxidation. This observation might have significant implications for the atmospheric fate of 1,2-ISOPOOH as the further oxidation pathways might be much different for photolysis and OH addition. For the lack of experimental data, the quantum yield was assumed to be one in both cases. As part of the light absorption was measured at a longer wavelength in the visible spectrum, this value could be smaller. Further work should be dedicated to this topic to substantiate the estimated absorption cross section and assess the atmospheric impact of ISOPOOH photolysis.
Additionally, the photolysis rate was calculated for conditions matching the parametrization of the master chemical mechanism (MCM) and the parameters l, m and n were optimized for Equation (1) [57], resulting in the parameters given below.
l = 0.00246014
m = 0.1530299
n = −0.17217075
3.2. Product Characterization following the 4,3-ISOPOOH Synthesis
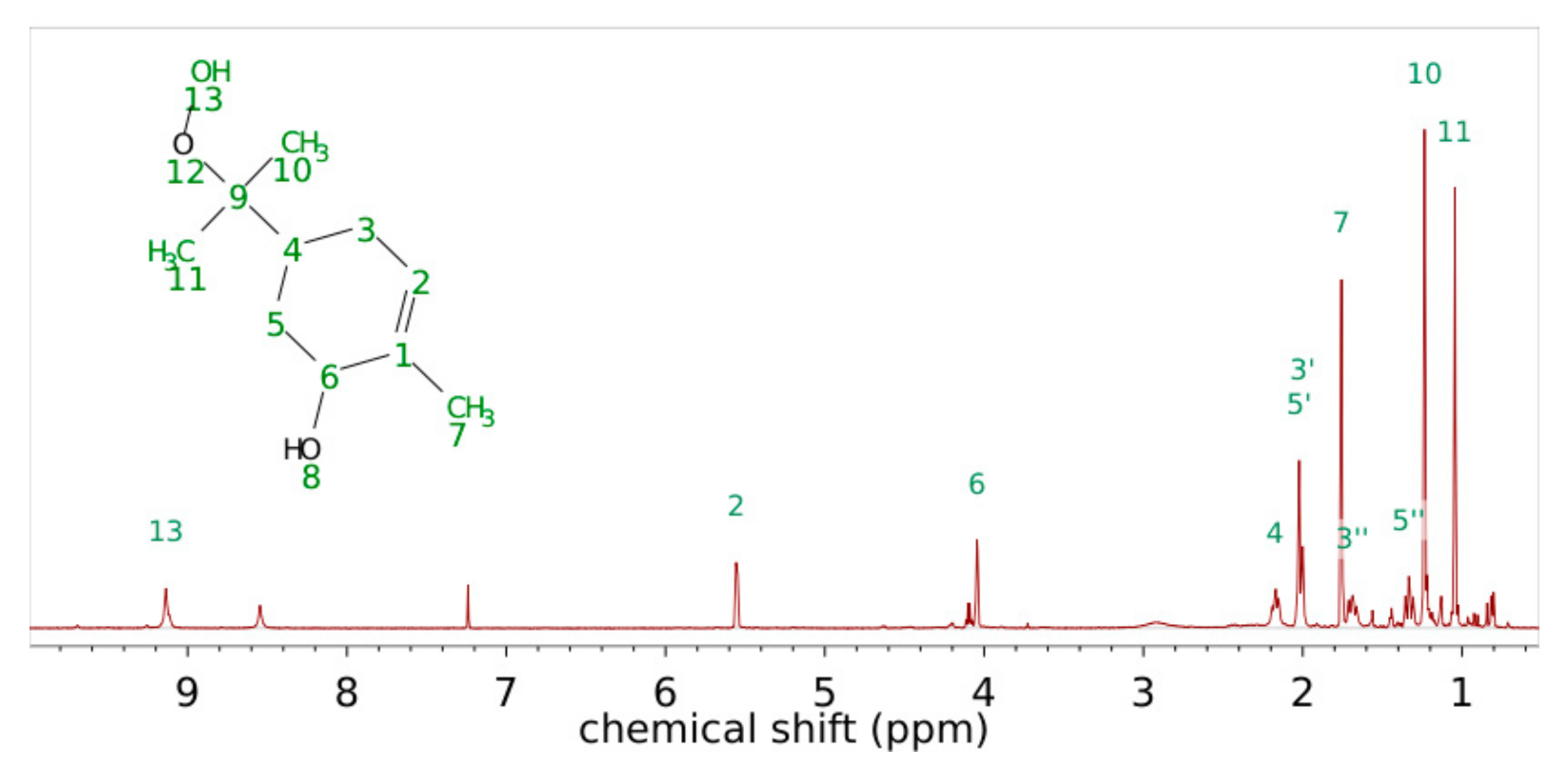
Similarly, the synthesis of 4,3-ISOPOOH was carried out from the corresponding isomer of isoprene epoxide. Since the precursor compound is not commercially available, synthesis was performed via a Johnson-Corey-Chaykowsky reaction according to Forrester et al. [58]. Notably, residual sodium hydroxide was washed out carefully during the reaction work-up to avoid a strong base being present during the epoxide ring opening. Even small amounts of residual sodium hydroxide decreased the product yield drastically. The conversion of the 1-epoxy-3-methyl-but-3-ene to the corresponding α-hydroxy-hydroperoxide was carried out similarly to the synthesis of 1,2-ISOPOOH, resulting in the clean product. Figure 5 shows the 1H-NMR spectrum with the peak assignments. The 13C, COSY, HMBC and HSQC spectra together with detailed peak assignments can be found in the supporting information Figures S6–S9 as well as Table S2.
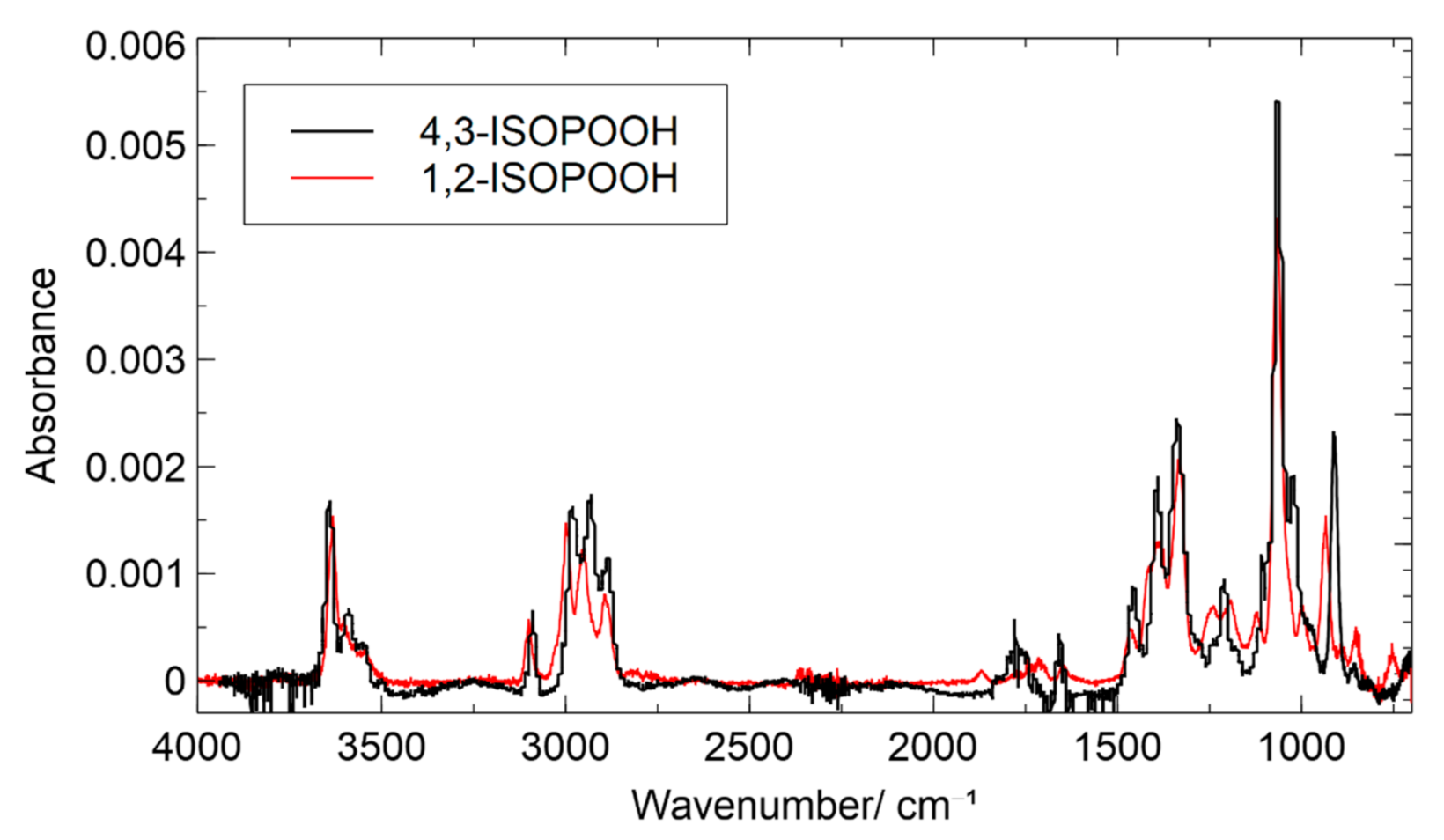
In comparison to the NMR spectrum of 1,2-ISOPOOH, the signal of the methyl group is shifted, and only one aliphatic signal of two proton equivalents could be observed, which is in good agreement with the molecular structure. The protons attached to the methylene carbon C1 show a similar chemical shift of 3.72 ppm and accordingly could contribute to the unresolved background of isoprene SOA (see Section 3.1 synthesis of 1,2-ISOPOOH). The NMR spectrum shows all reported signals that were previously reported by Dovrou et al. [45] with slightly different chemical shifts because of the use of CDCl3 as solvent. In the recorded spectrum, two low field signals are visible. The peak at 8.6 ppm can be assigned to the peroxy group of 4,3-ISOPOOH. The smaller peak at 7.8 ppm could correspond to the proton of the hydroxy group, as suggested for a previously reported NMR spectrum of ISOPOOH [43]. The unusual low field signal could indicate a strong hydrogen bond to the vicinal peroxy group. Such a peak, however, is not present in the spectrum of 1,2-ISOPOOH, which could be explained by the exchange of the proton with deuterium over time. However, since the NMR spectra of other hydroxy hydroperoxides [41] show only one low field signal, the signal was attributed to residual hydrogen peroxide, which was not completely removed during chromatographic purification. The fraction was calculated as below 4%, and based on other small visible NMR traces that we could not further identify, the purity of 4,3-ISOPOOH was estimated as greater than 95%. The gas-phase FTIR- and UV/VIS spectra were recorded and are presented here for the first time. Figure 6 shows the IR spectra of 1,2-ISOPOOH and 4,3-ISOPOOH overlaid. As both compounds are structurally very similar, both spectra feature similar patterns in the O-H stretch region as well as the C-H stretch region. 4,3-ISOPOOH, however, shows slightly higher absorptions at 2950 and 2890 cm−1. C-H bonds of sp2 hybridized carbon atoms show weaker absorption than C-H bonds on sp3 hybridized carbons. Since 4,3-ISOPOOH features one additional C-H bond on the unsaturated backbone, this observation is in good agreement with the molecular structure. The signals in the fingerprint region are also generally similar for both ISOPOOH isomers. The biggest difference is a shift in the absorption band previously attributed to the coupled stretching of the C-O-O bond. As the absorption peak for 1,2-ISOPOOH was at 935 cm−1, it is shifted to 915 cm−1 for 4,3-ISOPOOH, indicating a change in the bond strength of the peroxy group that is attached to a secondary carbon for 4,3-ISOPOOH and to a tertiary carbon for 1,2-ISOPOOH.
Notably, there is still a small carbonyl absorption band present in the spectrum, presumably originating from wall conversions of the compound resulting in bond cleavage and decomposition to carbonyl compounds [50]. To estimate the gas-phase concentration of 4,3-ISOPOOH, a flask containing the analytical standard was flushed for 3.5 h with the same flow of nitrogen as for 1,2-ISOPOOH. A total of merely 2.6 mg evaporated in this time period, yielding an average gas-phase concentration of 6.31 × 1013 molecule·cm−3, which is less than half the concentration observed for 1,2-ISOPOOH. This method can only give estimations for the gas-phase concentration, and further investigations of the phase transfer behavior of these compounds are necessary. A lower gas-phase concentration, however, might explain higher relative signals of carbonyl degradation products originating from similar wall effects. The UV/VIS spectrum in Figure S10 shows no visible differences compared to the spectrum of 1,2-ISOPOOH. The resulting PTR-ToFMS and NO3−-CI-APi-ToFMS spectra are shown in Figure S11.
3.3. α-Pinene Hydroxy Hydroperoxide Synthesis
The newly developed and improved method for the synthesis of α-hydroxy hydroperoxides was also applied to the most abundant monoterpene, α-pinene. TLC monitoring of the reaction mixture suggested a slower conversion rate. Hence the reaction time was extended to 1.5 h. A similar workup yielded a colorless oil in a smaller yield compared to isoprene-derived hydroxy hydroperoxides. Furthermore, the NMR investigation suggested a rearranged configuration of the hydroxy hydroperoxide. Briefly, the 1H spectrum shows a signal with a shift of 5.56 ppm, indicating the presence of a double bond in the product, as shown in Figure 7. As the spectrum shows one signal with a chemical shift at 4.04 ppm, the presence of a hydroxy group in the structure can be assumed.
The 13C, COSY and HSQC Spectra are shown in Figures S12–S14, respectively. On the basis of the C-H as well as the H-H coupling shown in the HMBC-spectrum in Figure S15, the structure of the resulting hydroxy hydroperoxide shown in Figure 7 was proposed.
Table 1 summarizes the peak assignments as well as the observed H-H and C-H coupling constants. The chemical shift of the carbon atoms C1 and C2 confirm the presence of a double bond in the molecular structure. Furthermore, one of the carbon atoms did not show any coupling to a hydrogen atom, suggesting the position at the methyl group C7. In contrast to the carbon atom C4, C2 did not show any coupling to the two remaining methyl groups C10 and C11. Thus, the original bicyclic α-pinene structure could be ruled out. Nevertheless, the chemical shifts of the carbon atoms C6 and C9 match the structure of a hydroxy hydroperoxide. Accordingly, the carbon atom C6 shows a coupling with the aliphatic proton H2 as well as with the remaining methyl group protons H7. Furthermore, in the HSQC spectrum, a coupling to the signal with the chemical shift of 4.06 could be observed, indicating the position of the hydroxy group in the vicinal position to the double bond.
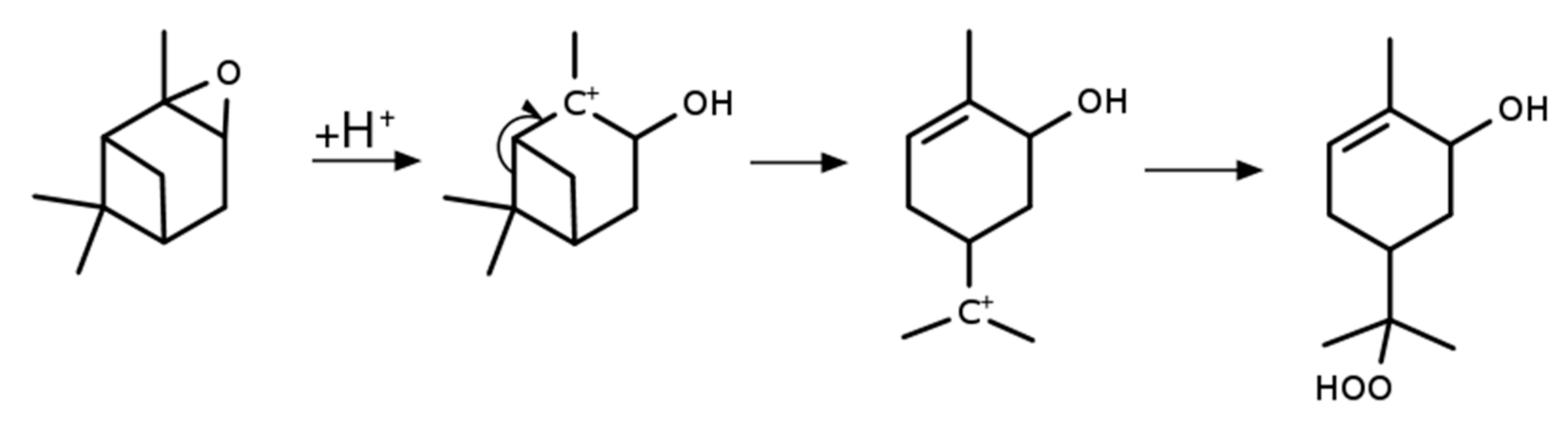
The catalytic ring opening of the epoxide during synthesis might result in a rearrangement during the reaction. The α-pinene bicyclic system with the additional three-membered ring of the epoxide might provide enough ring strain to drive this rearrangement even with the smallest amounts of protic solvents or reaction partners, as shown in Scheme 4.
Such ring-opening reactions also occur in the atmosphere, making the resulting organic peroxide of potential interest for atmospheric chemistry, although the formation pathway would include a bimolecular reaction of a peroxyl radical, which is formed after ring opening. The competitive pathway of intramolecular H-abstraction has been shown to occur very fast but is also highly dependent on the molecular conformation. The purity was determined from the 1H spectrum to be ≥87%, and the identified impurities were ethyl acetate with 7%, hydrogen peroxide with 3% and n-hexane with 2%. Figure S16 shows the PTR-ToFMS and NO3−-CI-APi-ToFMS spectra of α-pinene hydroxy hydroperoxide.
3.4. Chamber Characterization of Hydroxy Hydroperoxides
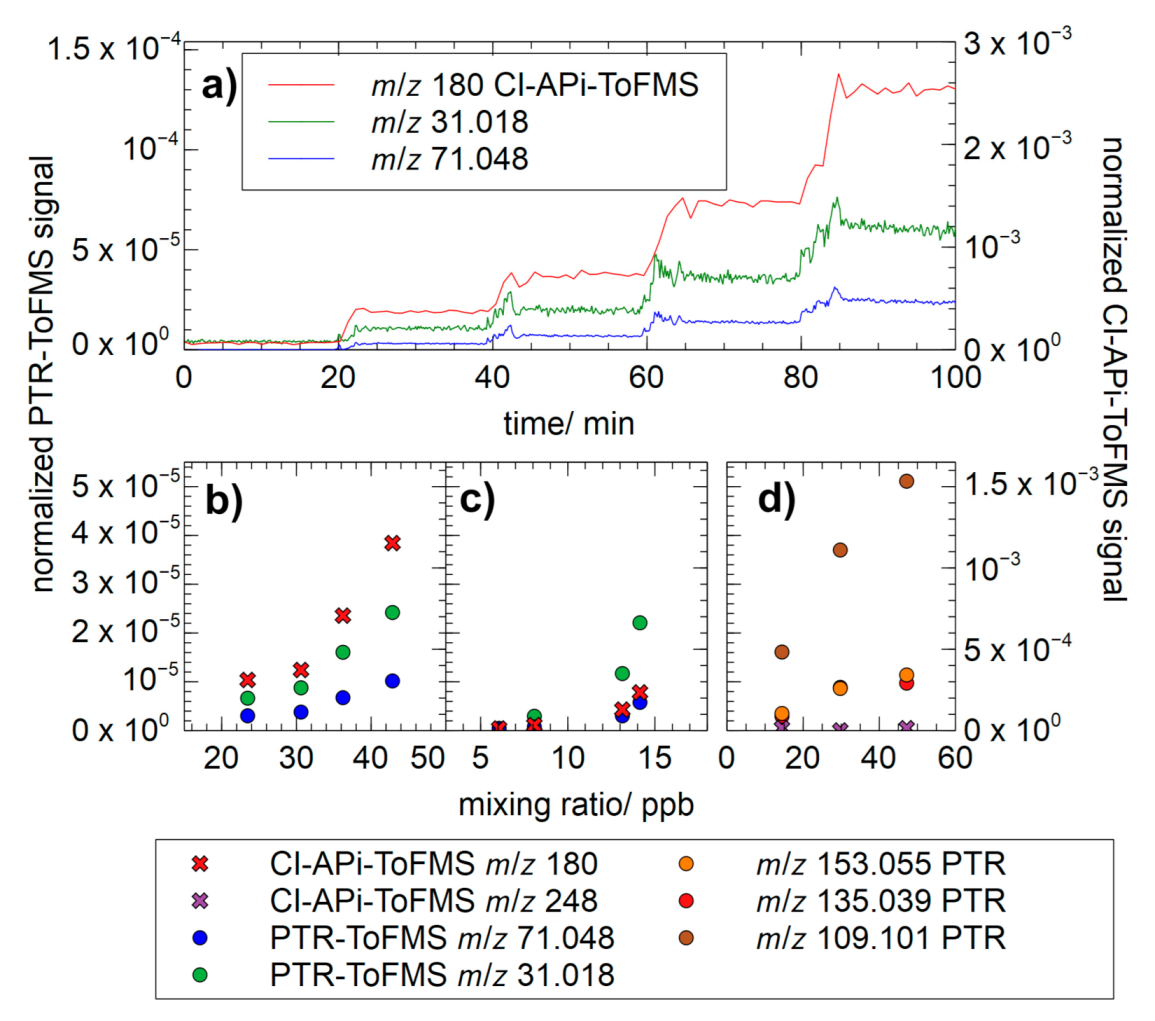
All synthesized standards were introduced to the chamber setup to investigate the response and fragmentation pattern of widely applied analytical methods, PTR-ToFMS and NO3−-CI-APi-ToFMS. Figure 8a shows an example of the calibration experiment of 1,2-ISOPOOH. The synthetic standard was placed in a flask and injected into the chamber with a gentle gas flow. All time series were corrected for chamber wall loss that was calculated as 4.16 × 10−5 s−1 for 1,2-ISOPOOH and as 4.56 × 10−5 s−1 for 4,3-ISOPOOH, respectively. The wall loss of the α-pinene hydroxy hydroperoxide was calculated as 1.06 × 10−5 s−1. Particle number concentrations were monitored with the MPSS to account for particle formation or occurrence, which was not observed through any of the experiments.
Figure 8b,c show the response of the PTR-ToFMS and the NO3−-CI-APi-ToFMS signals to the stepwise injection of 1,2-ISOPOOH and 4,3-ISOPOOH. For both isomers of ISOPOOH, an increase in the signal of the corresponding NO3−-cluster can be observed in the CI-APi-ToFMS. The calibration factor, however, is comparatively small with 4.44 × 10−5 and 2.47 × 10−5 ncps·ppbv−1, respectively. Consequently, the limit of detection (LOD) was calculated as (3σ, 1 min average) 0.70 ppbv for 1,2-ISOPOOH and 1.79 ppbv for 4,3-ISOPOOH. For lack of better standards, sulfuric acid is often used as a reference compound [59,60]. Even HOMs are expected to form less stable nitrate clusters compared to sulfuric acid. The calibration factor for sulfuric acid clusters of NO3−-CI-APi-ToFMS, however, usually ranges between 0.039 and 0.78 ncps·ppbv−1, which is at around three orders of magnitude higher [61]. This might indicate comparatively low cluster stability, which could be explained by the molecular structure of ISOPOOH compounds. Despite an O/C ratio of 0.6, they only possess two functional groups, probably resulting in less efficient nitrate cluster formation or less stable nitrate clusters. As a result, ISOPOOH might evade detection with NO3−-CI-APi-ToFMS depending on the MS-tuning parameters. The parameters used in this work are listed in Table S3. A strong influence of the tuning parameters of chemical ionization mass spectrometer (CIMS) has been reported before for the acetate ionization mode for the measurement of IEPOX by Budisulistiorini et al. [43]. In that study, the instrument response factor for 1,2-ISOPOOH was reported to be 9.95 × 10−5 ncps·ppbv−1, which is a factor of 2 higher compared to the present investigation. Another ionization method successfully applied for the measurement of ISOPOOH is the use of CF3O−. The use of tandem mass spectrometry also enabled the differentiation between the two isobaric compounds, ISOPOOH and IEPOX [23,56]. Vasquez et al. reported the coupling of gas chromatography (GC) to CF3O−-CIMS to distinguish between both compounds and other small isoprene-derived oxidation products [62]. Zhou et al. reported successful measurement of ISOPOOH with ammonium clustering atmospheric pressure chemical ionization tandem mass spectrometry (APCI-MS/MS). The mass loss of 51 Da from the parent ion m/z 136, corresponding to the loss of H2O2 + NH3, confirms the hydroperoxide group in the structure of ISOPOOH [63]. The PTRMS showed almost exclusively the signals of surface reaction and following α-bond cleavage after protonation, which is consistent with previous work [50,64,65]. For 1,2-ISOPOOH, methyl vinyl ketone is the main decomposition product, whereas for 4,3-ISOPOOH this process leads to the formation of primarily methacrolein. Both isobaric molecules can be detected at the mass-to-charge ratio m/z 71.048 with instrument responses of 3.97 × 10−7 ncps·ppbv−1 and 5.98 × 10−7 ncps·ppbv−1, respectively. Additionally, formaldehyde is formed during the process for both isomers with responses of 9.31 × 10−7 ncps·ppbv−1 and 2.47 × 10−6 ncps·ppbv−1, respectively.
For the case of the α-pinene hydroxy hydroperoxide, at the corresponding mass-to-charge ratio of the nitrate cluster of m/z 248, no increase in the signal in the CI-APi-ToFMS could be observed upon injection of the analytical standard as shown in Figure 8d. This might be attributed to even lower cluster stability, as the O/C ratio of the compound is lower (0.3) than ISOPOOH. Additionally, the functional groups are placed on opposite sides of the molecular structure, possible preventing efficient clustering with nitrate ions, making the use of other ionization methods necessary for efficient detection. The PTRMS, however, showed an increase in the signals at m/z 109.101, m/z 135.039 and m/z 153.055. The instrument responses were determined as 1.10 × 10−6 ncps·ppbv−1, 2.11 × 10−7 ncps·ppbv−1 and 2.48 × 10−7 ncps·ppbv−1, respectively.
4. Conclusions and Outlook
Within the present study, the two most abundant ISOPOOH isomers and an α-pinene-derived hydroxy hydroperoxide have been synthesized using an improved synthetic procedure. The structures of all three compounds have been confirmed by multiple NMR experiments, and further analytical data, namely gas-phase FTIR and UV/VIS spectra, were recorded. Using the TUV model, the atmospheric photolysis rate has been calculated from the determined UV/VIS absorption cross sections. Finally, the synthetic standards have been introduced to an atmospheric smog chamber setup and characterized with CI-APi-ToFMS and PTR-ToFMS, for which instrument response factors were determined. For both ISOPOOH isomers, signals of the corresponding nitrate cluster masses in the CI-APi-ToFMS were observed, and calibration factors as well as LODs were calculated.
The improved accessibility of isoprene-derived hydroxy hydroperoxides, as well as the provided analytical data, might enable further studies of their physicochemical properties. The multiphase chemistry of this compound class still needs further investigation to better assess different pathways of SOA formation and atmospheric oxidation. In recent years, some work has been dedicated to aqueous phase reactions of ISOPOOH [45,66,67]. However, for some key processes, such as phase transfer, experimental data are still widely missing. Given their high formation rates and influence on the oxidation budget of Earth’s atmosphere, detailed and comprehensive knowledge of these key intermediate products is needed. Furthermore, the photolysis rate of 1,2-ISOPOOH was found to be much higher than previously assumed. The implications of faster atmospheric photolysis, that might be competitive to OH oxidation in some cases, should be tested in a model study using the reported parametrization.
Similar to wall conversion pathways for chamber experiments that reportedly lead to serious underestimations of gas-phase concentrations of ISOPOOH isomers [50,64], the rapid decomposition of α-pinene-derived hydroxy hydroperoxides might lead to underestimation of these monoterpene reaction pathways. In order to better understand the atmospheric significance of these intermediate products, further studies using more sensitive analytical methods are necessary, as common techniques give unsatisfactory results. The improved access to the synthetic standard aims to enable focused future investigations of this topic.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/atmos13040507/s1, Figure S1: 13C-NMR spectrum of 1,2-ISOPOOH in CDCl3 at 600 MHz; Figure S2: COSY spectrum of 1,2-ISOPOOH in CDCl3 at 600 MHz; Figure S3: HSQC spectrum of 1,2-ISOPOOH in CDCl3 at 600 MHz; Figure S4: HMBC spectrum of 1,2-ISOPOOH in CDCl3 at 600 MHz; Table S1: Chemical shifts and coupling of NMR signals of 1,2-ISOPOOH.; Figure S5: NO3−-CI-APi-ToFMS spectrum (above) and PTR-ToFMS spectrum (below) of 1,2-ISOPOOH averaged over 5 min before the first and 10 min after the last injection.; Figure S6: 13C-NMR spectrum of 4,3-ISOPOOH in CDCl3 at 600 MHz; Figure S7: COSY spectrum of 4,3-ISOPOOH in CDCl3 at 600 MHz; Figure S8: HSQC spectrum of 4,3-ISOPOOH in CDCl3 at 600 MHz; Figure S9: HMBC spectrum of 4,3-ISOPOOH in CDCl3 at 600 MHz; Table S2: Chemical shifts and coupling of NMR signals of 4,3-ISOPOOH.; Figure S10: UV/VIS spectrum of 4,3-ISOPOOH, measured in a diluted gas stream at a mixing ratio of approximately 3.16 × 1013 molecule cm−3. Figure S11: NO3−-CI-APi-ToFMS spectrum (above) and PTR-ToFMS spectrum (below) of 1,2-ISOPOOH averaged over 5 min before the first and 10 min after the last injection.; Figure S12: 13C-NMR spectrum of alpha pinene hydroxy hydroperoxide in CDCl3 at 600 MHz; Figure S13: COSY spectrum of alpha pinene hydroxy hydroperoxide in CDCl3 at 600 MHz; Figure S14: HSQC spectrum of alpha pinene hydroxy hydroperoxide in CDCl3 at 600 MHz; Figure S15: HMBC spectrum of alpha pinene hydroxy hydroperoxide in CDCl3 at 600 MHz; Figure S16: NO3−-CI-APi-ToFMS spectrum(above and PTR-ToFMS spectrum (below) of 1,2-ISOPOOH averaged over 5 min before the first and 10 min after the last injection.; Table S3: TPS voltage settings for compact NO3−-CI-APi-ToFMS.
Author Contributions
Conceptualization, P.M., A.M. and H.H.; methodology, P.M. and O.B.; formal analysis, and investigation, P.M.; writing—original draft preparation, P.M.; writing—review and editing, P.M. and H.H.; resources and project administration, H.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work received funding from the European Union’s Horizon 2020 research and innovation programme through the EUROCHAMP-2020 Infrastructure Activity under grant agreement No 730997.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
We thank Jan Griebel for recording numerous NMR spectra. Furthermore, we thank Erik Hoffmann for the help with the photolysis rate calculation and Torsten Berndt for support during the recording of FTIR and UV/VIS spectra and helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest. The sponsors had no role in the design, execution, interpretation, or writing of this study.
References
- Kanakidou, M.; Seinfeld, J.H.; Pandis, S.N.; Barnes, I.; Dentener, F.J.; Facchini, M.C.; Van Dingenen, R.; Ervens, B.; Nenes, A.; Nielsen, C.J.; et al. Organic aerosol and global climate modelling: A review. Atmos. Chem. Phys. 2005, 5, 1053–1123. [Google Scholar] [CrossRef] [Green Version]
- Sindelarova, K.; Granier, C.; Bouarar, I.; Guenther, A.; Tilmes, S.; Stavrakou, T.; Müller, J.F.; Kuhn, U.; Stefani, P.; Knorr, W. Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years. Atmos. Chem. Phys. 2014, 14, 9317–9341. [Google Scholar] [CrossRef] [Green Version]
- Patra, P.K.; Krol, M.C.; Prinn, R.G.; Takigawa, M.; Mühle, J.; Montzka, S.A.; Lal, S.; Yamashita, Y.; Naus, S.; Chandra, N.; et al. Methyl Chloroform Continues to Constrain the Hydroxyl (OH) Variability in the Troposphere. J. Geophys. Res. Atmos. 2021, 126, 1–15. [Google Scholar] [CrossRef]
- Matsumi, Y.; Kawasaki, M. Photolysis of Atmospheric Ozone in the Ultraviolet Region. Chem. Rev. 2003, 103, 4767–4782. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, J.; Butler, T.M.; Crowley, J.N.; Dillon, T.J.; Fischer, H.; Ganzeveld, L.; Harder, H.; Lawrence, M.G.; Martinez, M.; Taraborrelli, D.; et al. Atmospheric oxidation capacity sustained by a tropical forest. Nature 2008, 452, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Barker, J.R.; Song, K. OH + Isoprene: A Direct Dynamics Study. Bull. Korean Chem. Soc. 2017, 38, 651–660. [Google Scholar] [CrossRef]
- Teng, A.P.; Crounse, J.D.; Wennberg, P.O. Isoprene Peroxy Radical Dynamics. J. Am. Chem. Soc. 2017, 139, 5367–5377. [Google Scholar] [CrossRef] [Green Version]
- Pratt, D.A.; Mills, J.H.; Porter, N.A. Theoretical Calculations of Carbon−Oxygen Bond Dissociation Enthalpies of Peroxyl Radicals Formed in the Autoxidation of Lipids. J. Am. Chem. Soc. 2003, 125, 5801–5810. [Google Scholar] [CrossRef]
- Peeters, J.; Nguyen, T.L.; Vereecken, L. HOx radical regeneration in the oxidation of isoprene. Phys. Chem. Chem. Phys. 2009, 11, 5935–5939. [Google Scholar] [CrossRef]
- Crounse, J.D.; Paulot, F.; Kjaergaard, H.G.; Wennberg, P.O. Peroxy radical isomerization in the oxidation of isoprene. Phys. Chem. Chem. Phys. 2011, 13, 13607–13613. [Google Scholar] [CrossRef] [Green Version]
- Berndt, T.; Hyttinen, N.; Herrmann, H.; Hansel, A. First oxidation products from the reaction of hydroxyl radicals with isoprene for pristine environmental conditions. Commun. Chem. 2019, 2, 21. [Google Scholar] [CrossRef]
- D’Ambro, E.L.; Moller, K.H.; Lopez-Hilfiker, F.D.; Schobesberger, S.; Liu, J.; Shilling, J.E.; Lee, B.H.; Kjaergaard, H.G.; Thornton, J.A. Isomerization of Second-Generation Isoprene Peroxy Radicals: Epoxide Formation and Implications for Secondary Organic Aerosol Yields. Env. Sci. Technol. 2017, 51, 4978–4987. [Google Scholar] [CrossRef]
- Jenkin, M.E.; Boyd, A.A.; Lesclaux, R. Peroxy Radical Kinetics Resulting from the OH-Initiated Oxidation of 1,3-Butadiene, 2,3-Dimethyl-1,3-Butadiene and Isoprene. J. Atmos. Chem. 1998, 29, 267–298. [Google Scholar] [CrossRef]
- Tyndall, G.S.; Cox, R.A.; Granier, C.; Lesclaux, R.; Moortgat, G.K.; Pilling, M.J.; Ravishankara, A.R.; Wallington, T.J. Atmospheric chemistry of small organic peroxy radicals. J. Geophys. Res. Atmos. 2001, 106, 12157–12182. [Google Scholar] [CrossRef]
- Orlando, J.J.; Tyndall, G.S. Laboratory studies of organic peroxy radical chemistry: An overview with emphasis on recent issues of atmospheric significance. Chem. Soc. Rev. 2012, 41, 6294–6317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndt, T.; Scholz, W.; Mentler, B.; Fischer, L.; Herrmann, H.; Kulmala, M.; Hansel, A. Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere. Angew. Chem. Int. Ed. 2018, 57, 3820–3824. [Google Scholar] [CrossRef]
- Wang, Y.; Mehra, A.; Krechmer, J.E.; Yang, G.; Hu, X.; Lu, Y.; Lambe, A.; Canagaratna, M.; Chen, J.; Worsnop, D.; et al. Oxygenated products formed from OH-initiated reactions of trimethylbenzene: Autoxidation and accretion. Atmos. Chem. Phys. 2020, 20, 9563–9579. [Google Scholar] [CrossRef]
- Tomaz, S.; Wang, D.; Zabalegui, N.; Li, D.; Lamkaddam, H.; Bachmeier, F.; Vogel, A.; Monge, M.E.; Perrier, S.; Baltensperger, U.; et al. Structures and reactivity of peroxy radicals and dimeric products revealed by online tandem mass spectrometry. Nat. Commun. 2021, 12, 300. [Google Scholar] [CrossRef]
- Vaughan, S.; Ingham, T.; Whalley, L.K.; Stone, D.; Evans, M.J.; Read, K.A.; Lee, J.D.; Moller, S.J.; Carpenter, L.J.; Lewis, A.C.; et al. Seasonal observations of OH and HO<sub>2</sub> in the remote tropical marine boundary layer. Atmos. Chem. Phys. 2012, 12, 2149–2172. [Google Scholar] [CrossRef] [Green Version]
- Madronich, S.; Calvert, J.G. Permutation reactions of organic peroxy radicals in the troposphere. J. Geophys. Res. Atmos. 1990, 95, 5697–5715. [Google Scholar] [CrossRef]
- Holzinger, R.; Sanhueza, E.; von Kuhlmann, R.; Kleiss, B.; Donoso, L.; Crutzen, P.J. Diurnal cycles and seasonal variation of isoprene and its oxidation products in the tropical savanna atmosphere. Glob. Biogeochem. Cycles 2002, 16, 22-1–22-13. [Google Scholar] [CrossRef]
- Crutzen, P.J.; Williams, J.; Pöschl, U.; Hoor, P.; Fischer, H.; Warneke, C.; Holzinger, R.; Hansel, A.; Lindinger, W.; Scheeren, B.; et al. High spatial and temporal resolution measurements of primary organics and their oxidation products over the tropical forests of Surinam. Atmos. Environ. 2000, 34, 1161–1165. [Google Scholar] [CrossRef]
- Paulot, F.; Crounse, J.D.; Kjaergaard, H.G.; Kürten, A.; St. Clair, J.M.; Seinfeld, J.H.; Wennberg, P.O. Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene. Science 2009, 325, 730. [Google Scholar] [CrossRef] [Green Version]
- Crounse, J.D.; McKinney, K.A.; Kwan, A.J.; Wennberg, P.O. Measurement of Gas-Phase Hydroperoxides by Chemical Ionization Mass Spectrometry. Anal. Chem. 2006, 78, 6726–6732. [Google Scholar] [CrossRef] [Green Version]
- Vereecken, L.; Peeters, J. Theoretical Study of the Formation of Acetone in the OH-Initiated Atmospheric Oxidation of α-Pinene. J. Phys. Chem. A 2000, 104, 11140–11146. [Google Scholar] [CrossRef]
- Berndt, T.; Richters, S.; Jokinen, T.; Hyttinen, N.; Kurten, T.; Otkjaer, R.V.; Kjaergaard, H.G.; Stratmann, F.; Herrmann, H.; Sipila, M.; et al. Hydroxyl radical-induced formation of highly oxidized organic compounds. Nat. Commun. 2016, 7, 13677. [Google Scholar] [CrossRef] [PubMed]
- Vereecken, L.; Muller, J.F.; Peeters, J. Low-volatility poly-oxygenates in the OH-initiated atmospheric oxidation of alpha-pinene: Impact of non-traditional peroxyl radical chemistry. Phys. Chem. Chem. Phys. 2007, 9, 5241–5248. [Google Scholar] [CrossRef] [PubMed]
- Peeters, J.; Vereecken, L.; Fantechi, G. The detailed mechanism of the OH-initiated atmospheric oxidation of α-pinene: A theoretical study. Phys. Chem. Chem. Phys. 2001, 3, 5489–5504. [Google Scholar] [CrossRef]
- Berndt, T. Peroxy Radical Processes and Product Formation in the OH Radical-Initiated Oxidation of alpha-Pinene for Near-Atmospheric Conditions. J. Phys. Chem. A 2021, 125, 9151–9160. [Google Scholar] [CrossRef]
- Xu, L.; Moller, K.H.; Crounse, J.D.; Otkjwr, R.V.; Kjaergaard, H.G.; Wennberg, P.O. Unimolecular Reactions of Peroxy Radicals Formed in the Oxidation of alpha-Pinene and beta-Pinene by Hydroxyl Radicals. J. Phys. Chem. A 2019, 123, 1661–1674. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Tsona, N.T.; Du, L. Relative Humidity Changes the Role of SO2 in Biogenic Secondary Organic Aerosol Formation. J. Phys. Chem. Lett. 2021, 12, 7365–7372. [Google Scholar] [CrossRef]
- Madronich, S.; Flocke, S. Theoretical Estimation of Biologically Effective UV Radiation at the Earth’s Surface. In Solar Ultraviolet Radiation, Proceedings of NATO ASI Series, Halkidiki, Greece, 2–11 October 1995; Zerefos, C.S., Bais, A.F., Eds.; Springer: Berlin/Heidelberg, Germany, 1997; pp. 23–48. [Google Scholar]
- Saunders, S.M.; Jenkin, M.E.; Derwent, R.G.; Pilling, M.J. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): Tropospheric degradation of non-aromatic volatile organic compounds. Atmos. Chem. Phys. 2003, 3, 161–180. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.W.; LaCount, R.B. The Chemistry of Ylids. VI. Dimethylsulfonium Fluorenylide—A Synthesis of Epoxides1. J. Am. Chem. Soc. 1961, 83, 417–423. [Google Scholar] [CrossRef]
- Corey, E.J.; Chaykovsky, M. Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis. J. Am. Chem. Soc. 1965, 87, 1353–1364. [Google Scholar] [CrossRef]
- Mutzel, A.; Poulain, L.; Berndt, T.; Iinuma, Y.; Rodigast, M.; Böge, O.; Richters, S.; Spindler, G.; Sipila, M.; Jokinen, T.; et al. Highly Oxidized Multifunctional Organic Compounds Observed in Tropospheric Particles: A Field and Laboratory Study. Environ. Sci. Technol. 2015, 49, 7754–7761. [Google Scholar] [CrossRef]
- Junninen, H.; Ehn, M.; Petäjä, T.; Luosujärvi, L.; Kotiaho, T.; Kostiainen, R.; Rohner, U.; Gonin, M.; Fuhrer, K.; Kulmala, M.; et al. A high-resolution mass spectrometer to measure atmospheric ion composition. Atmos. Meas. Tech. 2010, 3, 1039–1053. [Google Scholar] [CrossRef] [Green Version]
- Lindinger, W.; Hansel, A.; Jordan, A. On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) medical applications, food control and environmental research. Int. J. Mass Spectrom. Ion Processes 1998, 173, 191–241. [Google Scholar] [CrossRef]
- Birmili, W.; Stratmann, F.; Wiedensohler, A. Design of a Dma-Based Size Spectrometer for a Large Particle Size Range and Stable Operation. J. Aerosol Sci. 1999, 30, 549–553. [Google Scholar] [CrossRef]
- Riva, M.; Budisulistiorini, S.H.; Chen, Y.; Zhang, Z.; D’Ambro, E.L.; Zhang, X.; Gold, A.; Turpin, B.J.; Thornton, J.A.; Canagaratna, M.R.; et al. Chemical Characterization of Secondary Organic Aerosol from Oxidation of Isoprene Hydroxyhydroperoxides. Env. Sci. Technol. 2016, 50, 9889–9899. [Google Scholar] [CrossRef]
- Li, Y.; Hao, H.-D.; Wu, Y. Facile Ring-Opening of Oxiranes by H2O2 Catalyzed by Phosphomolybdic Acid. Org. Lett. 2009, 11, 2691–2694. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Zhang, Z.-H.; Li, T.-S. Efficient Conversion of Epoxides into β-Hydroperoxy Alcohols Catalyzed by Antimony Trichloride/SiO2. Synthesis 2008, 2008, 3314–3318. [Google Scholar] [CrossRef]
- Budisulistiorini, S.H.; Li, X.; Bairai, S.T.; Renfro, J.; Liu, Y.; Liu, Y.J.; McKinney, K.A.; Martin, S.T.; McNeill, V.F.; Pye, H.O.T.; et al. Examining the effects of anthropogenic emissions on isoprene-derived secondary organic aerosol formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee ground site. Atmos. Chem. Phys. 2015, 15, 8871–8888. [Google Scholar] [CrossRef] [Green Version]
- Pires, R.V.; Pessoa, L.M.B.; Sant’Anna, M.d.A.d.; Fainleib, A.; Nunes, R.d.C.P.; Lucas, E.F. Synthesis and characterization of isoprene oligomers to compare different production chemical processes. Polímeros 2019, 29, 1–9. [Google Scholar] [CrossRef]
- Dovrou, E.; Bates, K.H.; Rivera-Rios, J.C.; Cox, J.L.; Shutter, J.D.; Keutsch, F.N. Towards a chemical mechanism of the oxidation of aqueous sulfur dioxide via isoprene hydroxyl hydroperoxides (ISOPOOH). Atmos. Chem. Phys. 2021, 21, 8999–9008. [Google Scholar] [CrossRef]
- Zanca, N.; Lambe, A.T.; Massoli, P.; Paglione, M.; Croasdale, D.R.; Parmar, Y.; Tagliavini, E.; Gilardoni, S.; Decesari, S. Characterizing source fingerprints and ageing processes in laboratory-generated secondary organic aerosols using proton-nuclear magnetic resonance (1H-NMR) analysis and HPLC HULIS determination. Atmos. Chem. Phys. 2017, 17, 10405–10421. [Google Scholar] [CrossRef] [Green Version]
- Bates, K.H.; Crounse, J.D.; St Clair, J.M.; Bennett, N.B.; Nguyen, T.B.; Seinfeld, J.H.; Stoltz, B.M.; Wennberg, P.O. Gas phase production and loss of isoprene epoxydiols. J. Phys. Chem. A 2014, 118, 1237–1246. [Google Scholar] [CrossRef] [Green Version]
- Shreve, O.D.; Heether, M.R.; Knight, H.B.; Swern, D. Infrared Absorption Spectra of Some Hydroperoxides, Peroxides, and Related Compounds. Anal. Chem. 1951, 23, 282–285. [Google Scholar] [CrossRef]
- Brauer, C.S.; Blake, T.A.; Guenther, A.B.; Sharpe, S.W.; Sams, R.L.; Johnson, T.J. Quantitative infrared absorption cross sections of isoprene for atmospheric measurements. Atmos. Meas. Tech. 2014, 7, 3839–3847. [Google Scholar] [CrossRef] [Green Version]
- Bernhammer, A.-K.; Breitenlechner, M.; Keutsch, F.N.; Hansel, A. Technical note: Conversion of isoprene hydroxy hydroperoxides (ISOPOOHs) on metal environmental simulation chamber walls. Atmos. Chem. Phys. 2017, 17, 4053–4062. [Google Scholar] [CrossRef] [Green Version]
- Vacque, V.; Sombret, B.; Huvenne, J.P.; Legrand, P.; Suc, S. Characterisation of the O-O peroxide bond by vibrational spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1997, 53, 55–66. [Google Scholar] [CrossRef]
- Mohnhaupt, M.; Hagemann, H.; Perler, J.-P.; Bill, H.; Boukouvalas, J.; Rossier, J.-C.; Jefford, C.W. A Vibrational Study of Some 1,2,4-Trioxanes. Helv. Chim. Acta 1988, 71, 992–999. [Google Scholar] [CrossRef]
- Vaghjiani, G.L.; Ravishankara, A.R. Absorption cross sections of CH3OOH, H2O2, and D2O2 vapors between 210 and 365 nm at 297 K. J. Geophys. Res. Atmos. 1989, 94, 3487–3492. [Google Scholar] [CrossRef]
- Matthews, J.; Sinha, A.; Francisco, J.S. The importance of weak absorption features in promoting tropospheric radical production. Proc. Natl. Acad. Sci. USA 2005, 102, 7449. [Google Scholar] [CrossRef] [Green Version]
- Wennberg, P.O.; Bates, K.H.; Crounse, J.D.; Dodson, L.G.; McVay, R.C.; Mertens, L.A.; Nguyen, T.B.; Praske, E.; Schwantes, R.H.; Smarte, M.D.; et al. Gas-Phase Reactions of Isoprene and Its Major Oxidation Products. Chem. Rev. 2018, 118, 3337–3390. [Google Scholar] [CrossRef] [Green Version]
- St Clair, J.M.; Rivera-Rios, J.C.; Crounse, J.D.; Knap, H.C.; Bates, K.H.; Teng, A.P.; Jorgensen, S.; Kjaergaard, H.G.; Keutsch, F.N.; Wennberg, P.O. Kinetics and Products of the Reaction of the First-Generation Isoprene Hydroxy Hydroperoxide (ISOPOOH) with OH. J. Phys. Chem. A 2016, 120, 1441–1451. [Google Scholar] [CrossRef] [Green Version]
- Jenkin, M.E.; Saunders, S.M.; Pilling, M.J. The tropospheric degradation of volatile organic compounds: A protocol for mechanism development. Atmos. Environ. 1997, 31, 81–104. [Google Scholar] [CrossRef]
- Forrester, J.; Jones, R.V.H.; Preston, P.N.; Simpson, E.S.C. Efficient use of trimethylsulfonium methylsulfate as a reagent for the epoxidation of carbonyl-containing compounds. J. Chem. Soc. Perkin Trans. 1999, 22, 3333–3335. [Google Scholar] [CrossRef]
- Ehn, M.; Thornton, J.A.; Kleist, E.; Sipilä, M.; Junninen, H.; Pullinen, I.; Springer, M.; Rubach, F.; Tillmann, R.; Lee, B.; et al. A large source of low-volatility secondary organic aerosol. Nature 2014, 506, 476–479. [Google Scholar] [CrossRef]
- Kürten, A.; Rondo, L.; Ehrhart, S.; Curtius, J. Calibration of a Chemical Ionization Mass Spectrometer for the Measurement of Gaseous Sulfuric Acid. J. Phys. Chem. A 2012, 116, 6375–6386. [Google Scholar] [CrossRef]
- Bianchi, F.; Kurtén, T.; Riva, M.; Mohr, C.; Rissanen, M.P.; Roldin, P.; Berndt, T.; Crounse, J.D.; Wennberg, P.O.; Mentel, T.F.; et al. Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol. Chem. Rev. 2019, 119, 3472–3509. [Google Scholar] [CrossRef] [Green Version]
- Vasquez, K.T.; Allen, H.M.; Crounse, J.D.; Praske, E.; Xu, L.; Noelscher, A.C.; Wennberg, P.O. Low-pressure gas chromatography with chemical ionization mass spectrometry for quantification of multifunctional organic compounds in the atmosphere. Atmos. Meas. Tech. 2018, 11, 6815–6832. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Rivera-Rios, J.C.; Keutsch, F.N.; Abbatt, J.P.D. Identification of organic hydroperoxides and peroxy acids using atmospheric pressure chemical ionization–tandem mass spectrometry (APCI-MS/MS): Application to secondary organic aerosol. Atmos. Meas. Tech. 2018, 11, 3081–3089. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Rios, J.C.; Nguyen, T.B.; Crounse, J.D.; Jud, W.; St. Clair, J.M.; Mikoviny, T.; Gilman, J.B.; Lerner, B.M.; Kaiser, J.B.; de Gouw, J.; et al. Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: Observational bias in diagnosing pristine versus anthropogenically controlled atmospheric chemistry. Geophys. Res. Lett. 2014, 41, 8645–8651. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.J.; Herdlinger-Blatt, I.; McKinney, K.A.; Martin, S.T. Production of methyl vinyl ketone and methacrolein via the hydroperoxyl pathway of isoprene oxidation. Atmos. Chem. Phys. 2013, 13, 5715–5730. [Google Scholar] [CrossRef] [Green Version]
- Fang, T.; Lakey, P.S.J.; Rivera-Rios, J.C.; Keutsch, F.N.; Shiraiwa, M. Aqueous-Phase Decomposition of Isoprene Hydroxy Hydroperoxide and Hydroxyl Radical Formation by Fenton-like Reactions with Iron Ions. J. Phys. Chem. A 2020, 124, 5230–5236. [Google Scholar] [CrossRef]
- Dovrou, E.; Rivera-Rios, J.C.; Bates, K.H.; Keutsch, F.N. Sulfate Formation via Cloud Processing from Isoprene Hydroxyl Hydroperoxides (ISOPOOH). Environ. Sci. Technol. 2019, 53, 12476–12484. [Google Scholar] [CrossRef]
Scheme 1.
Major first-generation reaction pathways for isoprene OH oxidation.

Scheme 2.
Proposed first OH oxidation steps of α-pinene with assigned branching ratios.

Scheme 3.
General synthetic procedure for the synthesis of hydroxy hydroperoxides. In the following, the respective working steps are described.
Scheme 3.
General synthetic procedure for the synthesis of hydroxy hydroperoxides. In the following, the respective working steps are described.

Figure 1.
1H NMR Spectrum of 1,2-ISOPOOH in CDCl3 at 600 MHz with peak assignments.

Figure 2.
Gas phase FTIR Spectrum of 1,2-ISOPOOH.

Figure 3.
Gas phase UV/VIS Spectrum of 1,2-ISOPOOH, measured in a diluted gas stream at a mixing ratio of approximately 6.61 × 1013 molecules·cm−3.
Figure 3.
Gas phase UV/VIS Spectrum of 1,2-ISOPOOH, measured in a diluted gas stream at a mixing ratio of approximately 6.61 × 1013 molecules·cm−3.

Figure 4.
Photolysis rate of 1,2-ISOPOOH over the course of a day on 21 March at latitudes between 0° and 30°.
Figure 4.
Photolysis rate of 1,2-ISOPOOH over the course of a day on 21 March at latitudes between 0° and 30°.

Figure 5.
1H-NMR spectrum of 4,3-ISOPOOH in CDCl3 at 600 MHz.

Figure 6.
Gas-phase FTIR spectrum of 4,3-ISOPOOH, combined with the FTIR spectrum of 1,2-ISOPOOH (red line).
Figure 6.
Gas-phase FTIR spectrum of 4,3-ISOPOOH, combined with the FTIR spectrum of 1,2-ISOPOOH (red line).

Figure 7.
1H-NMR spectrum of α-pinene hydroxy hydroperoxide at 600 MHz.

Scheme 4.
Proposed mechanism of the ring opening during the synthesis of the α-pinene derived α-hydroxy hydroperoxide.
Scheme 4.
Proposed mechanism of the ring opening during the synthesis of the α-pinene derived α-hydroxy hydroperoxide.

Figure 8.
(a) Calibration experiment of 1,2-ISOPOOH with stepwise injection of the analytical compound over a gentle gas flow. Panels (b,c) show the response-curve of both the corresponding APi-ToFMS peak and PTRMS fragments for 1,2-ISOPOOH (b) and 4,3-ISOPOOH (c), respectively. Similarly, the response of these instruments for the α-pinene-derived hydroxy hydroperoxide is shown in panel (d).
Figure 8.
(a) Calibration experiment of 1,2-ISOPOOH with stepwise injection of the analytical compound over a gentle gas flow. Panels (b,c) show the response-curve of both the corresponding APi-ToFMS peak and PTRMS fragments for 1,2-ISOPOOH (b) and 4,3-ISOPOOH (c), respectively. Similarly, the response of these instruments for the α-pinene-derived hydroxy hydroperoxide is shown in panel (d).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Chemical shifts and coupling of NMR signals.
| Carbon Atom Number | Shift of Hydrogen Atom in 1H-NMR/ppm | Shift of Carbon Atom in 13C-NMR/ppm | H-H Coupling COSY Coupling Constants J in Brackets (from 1H) | C-H Coupling HMBC |
|---|---|---|---|---|
| 1 | 134.22 | C1-H6; C1-H5′; C1-H7 | ||
| 2 | 5.56 | 125.85 | H2-H7; H2-H3′ (3Hz) | C2-H6; C2-H7 |
| 3 | 3′ 2.00 3″ 1.69 | 27.60 | H3′-H3″H3′-H2 (3 Hz); H3′-H4 H3″-H3′ (14.4 Hz); H3″-H4 (14.4 Hz) | C3-H2; C3-H5’ |
| 4 | 2.17 | 33.49 | H4-H3’ (14.4 Hz); H4-H3″; H4-H5′; H4-H5″ (13.5 Hz) | C4-H3′; C4-H3″; C4-H5′; C4-H5″, C4-H10; C4-H11 |
| 5 | 5′ 2.02 5″ 1.33 | 31.97 | H5′-H4, H5’-H5″ (13.6 Hz) H5″-H4 (13.6 Hz); H5″-H5′ (13.6 Hz); H5″-H6 | |
| 6 | 4.04 | 69.03 | H6-H5″, H6-H7 | C6-H2; C6-H5′; C6-H7 |
| 7 | 1.76 | 21.00 | H7-H6; H7-H3′ | |
| 9 | 84.45 | C9-H10; C9-H11 | ||
| 10 | 1.23 | 22.91 | C10-H11 | |
| 11 | 1.04 | 20.37 | C11-H10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mettke, P.; Mutzel, A.; Böge, O.; Herrmann, H. Synthesis and Characterization of Atmospherically Relevant Hydroxy Hydroperoxides. Atmosphere 2022, 13, 507. https://doi.org/10.3390/atmos13040507
AMA Style
Mettke P, Mutzel A, Böge O, Herrmann H. Synthesis and Characterization of Atmospherically Relevant Hydroxy Hydroperoxides. Atmosphere. 2022; 13(4):507. https://doi.org/10.3390/atmos13040507
Chicago/Turabian StyleMettke, Peter, Anke Mutzel, Olaf Böge, and Hartmut Herrmann. 2022. "Synthesis and Characterization of Atmospherically Relevant Hydroxy Hydroperoxides" Atmosphere 13, no. 4: 507. https://doi.org/10.3390/atmos13040507
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.