
Abstract
Esophageal cancer (EC) mostly affects the elderly population and is frequently diagnosed at an advanced stage. Self-expanding metal stents (SEMS) are the most popular mode of palliation, but they are associated with reocclusion caused by tumor growth. To overcome this problem, docetaxel (DTX)-loaded polyurethane formulations were prepared for stent application. The films were evaluated against the cancer cell lines, OE-19 and OE-21, and normal esophageal cell line Het-1A. The DTX and the formulations were evaluated in vitro for the cytotoxicity and in vivo in nude mice. It was found that DTX and the formulations have a weak activity against the EC cell lines and an even weaker activity against Het-1A cell line. Preliminary in vivo studies showed skin toxicity in nude mice necessitating modification of the formulation. Reevaluation in a mouse xenograft model resulted in toxicity at high dose formulations while the low dose formulation exhibited modest advantage over commercial IV formulation; however, there was no significant difference between the commercial IV and blank formulation. DTX combination with an anti-cancer agent having complementary mode of action and non-overlapping toxicity could yield better outcome in future.
Similar content being viewed by others


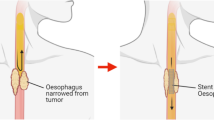
INTRODUCTION
Esophageal cancer (EC) is a leading cause of cancer-related deaths. Poor prognosis accompanied by late diagnosis has made EC treatment challenging. There are two main histological types of EC, squamous cell carcinoma (SCC) and adenocarcinoma (ACA). SCC comprises 90% of all the cases worldwide and is more prevalent in Eastern Africa, Middle East, and Asian countries. However, the incidence of ACA has increased over the last three decades and it is the predominant histological type found in the Western population including Western Europe, USA, and Australia (1).
Palliation is the only option for more than 50% of the patients diagnosed for EC (2). Furthermore, most of the patients are elderly (>65 years) with severe comorbidities; hence, the aim of palliation therapy is to improve the quality of life of the patients. Palliative stenting, laser therapy, photodynamic therapy, dilation, tumor ablation by electrocautery, and external beam irradiation are some of the palliation options, but stenting (2,3) using self-expanding metallic stents (SEMS) is the most commonly used therapy for the palliation of advanced EC. SEMS come in covered, partially covered, and uncovered forms. Of these, partially covered esophageal stents are preferred for palliation (3). But in the case of covered stents, 4–18% of cases experience tumor overgrowth at the ends of the stent (4).
Docetaxel (DTX) is an anticancer drug used in the treatment of EC but systemic use of DTX is associated with neurotoxicity, musculoskeletal toxicity, and neutropenia. In addition, currently marketed formulations (Taxotere® and Duopafei®) are implicated in hypersensitivity reactions necessitating glucocorticoid administration (5). Development of a localized delivery system would avoid the side effects arising from the formulation as well as drug. DTX-loaded PurSil®AL20 (silicone modified polyurethane polymer, PUS) films were prepared as a delivery system for stent application with the aim of preventing stent occlusion. These films were prepared in a bilayer configuration using DTX-loaded and blank film. Bilayer formulations have been shown to provide sustained unidirectional release over a month (6). In the present investigation, the formulations were tested for in vitro activity in cell types representing EC from both histological types, viz, OE-19 (ACA) and OE-21 (SCC). Cytotoxicity of DTX was also determined on the Het-1A cell line which is representative of normal esophageal epithelial cells. In vivo evaluation of the bilayer formulation and a commercial IV formulation (Duopafei®) was performed in mouse xenograft model.
METHODS
Formulation Preparation
DTX-loaded PU films were prepared by the solvent casting method described by Shaikh et al. (6). In brief, appropriate amount of PUS and DTX were dissolved in tetrahydrofuran (THF) and cast on a Petri plate. Blank and DTX-loaded formulations were then agglutinated using THF for making bilayer films. The following film formulations were prepared for in vitro evaluation (Formulation 1–4, Table I). For each thickness group (drug-loaded film) one high and one low dose were evaluated.
Formulation Modification
Formulations 3 and 4 showed sustained release as compared to formulations 1 and 2 (6). Considering the benefit of long-term activity against the tumors, formulation 3 and 4 were selected for initial in vivo studies. However, in preliminary studies in nude mice, DTX-related skin toxicities were observed. It was hypothesized that the release of DTX from the backside of the film is causing these toxicities. Hence, modification in the existing formulation was performed (Table I). Formulations 1 and 2 (Table I) were modified to make formulations 1m and 2m. DTX is soluble in THF and can cause its diffusion into the backing layer during film attachment. So, modified films were agglutinated using double-sided tape (3M™ Double coated tape polypropylene film with synthetic rubber adhesive) instead of THF to avoid backside release. Though use of a tape would have sufficed for the purpose, an additional PUS backing layer was used, to avoid film separation in vivo under wet condition and also to rule out contact of a new material (tape) in the advance stage of the study.
Characterization of the Modified Formulation
Formulations 1m and 2m (square piece of a 1-cm2 area, n = 3) were characterized as described previously (6). Physical characterization included weight and thickness variation. HPLC method was used for the assay and release sample analysis was as described by Shaikh et al. (6). Briefly, Zorbax Eclipse XDB C18 column (4.6 × 150-mm dimensions and 3.5-μm particle size) was used on a Shimadzu instrument equipped with DGU-20AS degasser, LC-20AD liquid chromatograph, SIL-20A HT autosampler and SPD M20A diode array detector. Isocratic gradient of ammonium acetate buffer (0.02M, pH 5) and acetonitrile at 43:57 ratio was run for 10 min. DTX release from the backside and in vitro release was performed in a pH 6 phosphate buffer with 0.1% Tween 80 at 37°C. For determining the backside release of DTX, a Franz diffusion cell (PermeGear, Inc., Bethlehem, PA 18015, 5 mL receptor volume) was used. Drug release was determined from the drug loaded and backside by exposing an appropriate side of the film to the receptor compartment. For in vitro release (15 mL release media), the backside was covered by a 3-mm-thick acrylate sheet to completely rule out the drug release from the backside. Cumulative release obtained from this study was then fitted in the solution provided by Fick’s second law of diffusion (Eq. 1). This data was also fitted in first order kinetic equation to determine rate constant as described previously (6). For both, the Franz diffusion study and in vitro release study, release media was completely replenished at each time point to maintain sink conditions.
Solubility of DTX in RPMI Medium
RPMI 1640 media with 10% fetal bovine serum (FBS) was incubated along with an excess amount of DTX at 37°C over 24 h. Samples were filtered through a PTFE filter (0.22 μm).
Cell Culture Maintenance
OE-19 (ACA) and OE-21 (SCC) EC cells lines (obtained from the Peter MacCallum Cancer Centre, Melbourne) were grown on RPMI 1640 medium (Life Technologies) with 10% FBS and 0.1% antibiotic-antimycotic at 37°C in the presence of 5% CO2. The Het-1A cell line was grown on LHC-9 medium at 37°C in the presence of 5% CO2.
Cell Viability Assay
Cytotoxicity was determined using the colorimetric 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay. Both, the DTX solution and the film formulations were evaluated for cytotoxicity.
DTX (DMSO solution) cytotoxicity assays were performed traditionally, in a 96-well plate. After 24 h of treatment with the drug, the cell cultures were incubated for 4 h at 37°C in the presence of 0.05 mg/ml MTT reagent solution prepared in phosphate buffer saline (PBS) and RPMI medium. Metabolically active cells convert MTT into purple formazan crystals. After the incubation period, wells were emptied and DMSO was added to each well to dissolve formazan and absorbance was measured at 570 nm.
For the films, non-cumulative cytotoxicity (7) was determined on day 1 and day 10 or day 11. Assays were performed in 24-well plates using a transwell (ThinCert™, 8 μm pore size, inner diameter 8.4 mm, Fig. 1) to support the film formulation during the test. Cells were seeded at 2 × 104 cells per well in a 24-well plate. Film formulations (0.5 × 0.5 cm2) were placed into a transwell (ThinCert™, 8 μm pore size, inner diameter 8.4 mm) aseptically. After performing cytotoxicity assay for 24 h at day 1 time point, films were kept in a new culture plate (24 wells) with culture medium (0.5 mL) to maintain release gradient. Media was changed every alternate day till day 10, after which films were again transferred for assay to a 24-well plate previously seeded with cells on day 10 or 11. A MTT assay was performed at both time points for cytotoxicity determination as described above.

Schematic presentation of the Transwell setting used for cytotoxicity assessment
LDH Release Assay
Cytotoxicity was also determined using the LDH (Lactate dehydrogenase) kit (Roche Applied Sciences) as per the manufacturer’s instruction. The test was performed in a transwell as described above (cell viability assay). Cytotoxicity was calculated by determining the LDH leakage caused due to treatment induced cell membrane damage. Untreated cells were used as a negative control (low control) and 1% v/v Triton X-100 solution in culture media was used as a positive control (high control). After 24 h of treatment (n = 3), culture media were analyzed along with controls and absorbance was measured in a microplate reader (Corona Electric) at 492 nm using 620 nm as a reference wavelength.
In Vivo Evaluation of the Formulations
In vivo films (1-cm2 area) were evaluated in female BALB/c-nu mice (16–20 g, 35–41-day old). This study was conducted in the School of Pharmacy, Shandong University as per the approval granted by the Animal Ethics Committee of Shandong University (ECAESDUSM). Preliminary studies on formulation 3 and 4 were performed (results not shown) and it was found that animals displayed dose-dependent skin toxicities and these toxicities were absent in a blank formulation group. Hence, formulations were modified and next round of studies were performed using formulations 1m and 2m.
Xenograft tumors were developed by injecting 1 × 107 KYSE30 human SCC cells suspension subcutaneously into the mice axilla. Tumors were dissociated (after reaching a 1-cm3 volume) and implanted into 25 nude mice. Tumors were allowed to grow up to 100 mm3 before treatment.
The study was divided into five groups (n = 5 each): (1) negative control, (2) single dose at 5 mg/kg IV group (Duopafei®, IV formulations were provided by Qilu Hospital Shandong Province, China), (3) blank formulation, (4) formulation 1m, and (5) formulation 2m. Films (1-cm2 area, DTX-loaded side facing tumor) were implanted at the right flank over the tumor by making a small incision of 0.5–1-cm length. Penicillin (8000 U per animal) was administered intramuscularly for the next 5 days to prevent infection. Every alternate day total body weights and tumor volumes were measured over 12 days. Tumor volumes were calculated by using V = 1/2(length × width ^ 2) formula. At the end of study, mice were sacrificed and tumors were removed and weighed.
Statistical Analysis
Results are presented as mean ± standard deviation. All statistical analyses were either performed using paired student’s t test or one-way ANOVA with LSD as a post hoc test.
RESULTS
Film Formulation Characterization
The surface dose density of formulations 1m and 2m was 514.71 and 94.82 μg cm-2, respectively (Table II). DTX loading of the formulations was in close agreement with our previously published studies (6).
The modified formulations were first characterized for the release from the backside using a Franz diffusion cell. It was found that on day 14 release was about 17.5-fold higher from the drug-loaded side compared to the backside (Fig. 2a).

Cumulative amount release from the a Franz diffusion cell study showing release from DTX-loaded front side (primary Y axis) and blank back side (secondary Y axis) of the formulation 1m and b in vitro release from modified formulations 1m and 2m
More than 90% of the DTX released over 20 days (Fig. 2b) in an in vitro release test from both formulations 1m and 2m. The diffusion coefficient for formulations 1m and 2m was found to be 5.44E–10 ± 5.7E–12 and 4.38E–10 ± 0.24E–11, respectively. The first order release rate constant for the formulation 1m and 2m were 0.131 ± 0.024 and 0.138 ± 0.072 d -1, respectively.
DTX Solubility in Culture Media
RPMI 1640 media with 10% FBS solubilizes 32.1 ± 3.3 μg/mL (39.6 μM) of DTX.
Cytotoxicity Assay
A stock solution of DTX in DMSO was tested on the Het-1A, OE-19, and OE-21 cell lines. It was observed that (Fig. 3) the activity of DTX was in the order of OE-21 > OE-19 > Het-1A. IC50 (concentration to cause 50% of cell death) values for OE-19 and OE-21 were calculated as 221.1 and 54.16 μM, respectively. In the case of Het-1A at the highest concentration (250 μM), cell viability was reduced by only 15% compared to the control.

MTT assay results for the DTX activity against esophageal epithelial cells (Het-1A) and esophageal cancer (OE-19 and OE-21) cells after 24 h of exposure
Formulation Activity In Vitro
In vitro, the film formulations have been shown to sustain release over a month (6). A non-cumulative cytotoxicity assay was found suitable for activity determination, though this assay does not allow long duration of testing but allows different time intervals during release period. This is especially helpful since film formulations show a biphasic release from day (D) 1-D3 and from D6 until end of the release (6). After D6, the release rate became steady. So, the formulations were screened for a period of 24 h at D0 and D10 or D11 time point to capture both phases of release. The release on D0–D1 was higher as compared to D10–D11 or D11–D12. So, the Het-1A cell lines were only tested at D0 for 24 h to check the cytotoxicity of DTX in a normal cell line. It was found that the Het-1A cell line showed insignificant reduction in cell viability after 24 h of treatment (Fig. 4). It was also found that DTX-eluting films show about 10% cytotoxicity in OE-19 cell lines when the activity was compared with the blank film. It was observed that all the formulations except formulations 1 and 4 show statistically insignificant difference at D10 and D0. Unexpectedly, OE-21 cell lines though having a lower IC50 value, did not show a significant cytotoxicity as compared to blank film.

MTT and LDH activity assay for the film formulation on Het-1A, OE-19, and OE-21 cell line. D stands for the day of activity testing and TXT for Triton X positive control sample. The number sign indicates the paired t test results at p < 0.05 between control and blank film.The asterisk indicates the paired t test results at p < 0.05 between blank film and formulations
LDH assay results show that LDH release was significantly lower for blank formulation than the positive control in all the cell lines (Fig. 4). Also, there was no significant difference between the formulations and the blank film for any of the cell lines.
In Vivo Evaluation of the Formulation
As explained earlier, formulations 3 (∼534-μg DTX) and 4 (∼99-μg DTX) were evaluated in a preliminary in vivo study. The toxicity was attributed mainly to DTX since the effect of implantation was dose dependent. Modified formulations 1m (∼514-μg DTX) and 2m (∼94-μg DTX) were then evaluated in vivo. Even though modification was performed to avoid skin toxicity, formulation 1m was found toxic and, hence, was discontinued on day 2. However, blank and formulation 2m were safe and studied for 12 days.
The weight change and the tumor volume progression are shown in Fig. 5a, b, respectively. Overall, no significant difference was observed in the body weight of the animals over the course of the study. The negative control, blank formulation, and the 5-mg/kg IV formulation group did not show any significant difference in tumor volume at any point during the study. The difference in tumor volume for formulation 2m was statistically significant (*p < 0.05) on day 2, 8, and 10 compared with the negative control sample. Formulation 2m also showed a statistical difference (#p < 0.05) on day 8 compared to the 5-mg/kg IV formulation. On day 12, no significant difference between any of the controls or treatment groups was observed.

Total body weight (a) and the tumor volume changes (b) during the study.The asterisk indicates the statistically significant difference (p < 0.05) between negative control and formulation 2m, the number sign indicates the statistically significant difference (p < 0.05) between 5-mg/kg IV group and formulation 2m group
At the end of the study, mice were sacrificed and tumors were weighed (Fig. 6a). As expected from the tumor volume analysis, no significant difference was observed in tumor weights or in percent inhibition rate (Fig. 6b). But trend wise, formulation 2m performed better as compared to the 5-mg/kg IV and blank formulations.

Mice and the tumor images (a). Tumor weight along with the tumor inhibition rate (%) at the end of the study (b)
DISCUSSION
In Vitro Activity
Cytotoxicity assay results indicated that DTX is active in order of OE-21 > OE-19 > Het-1A. This order follows the mitotic index of the cell lines. OE-21 is the fastest while Het-1A is the slowest growing cell line. DTX binds to microtubule and stabilizes the tubulin structure during mitosis. The above results confirm the earlier findings that DTX acts on actively dividing cells by arresting the cells in G2/M phase (8).
When formulation activity was studied, it was found that the OE-19 cell line showed significant cell killing compared to blank films, while in the case of OE-21 cells, no significant activity was observed. Though this was an unexpected observation, the results were found reproducible. Formulations 1 and 2 have higher in vitro DTX release compared to formulations 3 and 4 (6) due to the difference in the thickness. However, no significant difference was observed in cytotoxicity between these two film types for the OE-19 cell line. This could be due to solubility limitation which is covered in detail in the following paragraph. As for OE-21 cells, about 30% cell killing was observed but the results were not statistically significant when compared to the blank sample. Further experiments are needed to determine reasons for this observation.
Limited activity in vitro could stem from the fact that the solubility of DTX in the culture media is less (∼32 μg/mL or 39.6 μM). For assays, about 200–700 μL of media (depending on the assay) was used, which means the maximum amount of DTX that would dissolve (22.7 μg/mL or 28.1 μM) is less than the IC50. In addition, the tests were performed under static conditions (without shaking), which in turn reduces the amount of DTX released from the film formulation.
In the case of the LDH assay, there was no significant difference between the blank and the formulations for both OE-19 and OE-21, indicating insignificant damage to cell membrane over the 24 h of the study.
Modified Formulation and the In Vivo Activity
The first order rate constant and the diffusion coefficients of the modified formulations are within experimental variations when compared with our previous findings (6). Compared to the original films (biphasic release), the modified films show single phase release and hence whole profiles were fitted for calculating first order rate constant and diffusion coefficient. Previously, bilayer films were prepared by agglutination using THF which causes redistribution of DTX in backing layer (DTX is soluble in THF). This causes a decrease in release (second phase of slower release) as the diffusion front moves into the film (leading to biphasic release). This effect was not observed in the modified films causing single phase of release since the agglutination was performed using double-sided tape.
For formulation 1 it was found previously that the difference between the front side and backside release was 22-fold. In the current investigation, a 17.5-fold difference was observed. So, using a double-sided tape to stick the films does not change the backside release significantly (Fig. 2), and this could be due to the faster release observed from the backside of formulation 1m. Nonetheless, the benefit of using the modified formulation was evident in in vivo study. The amount of DTX in case of formulations 4 and 2m is the same. Also, the release rate is slower in case of formulation 4 compared to formulation 2m. However, in the case of formulation 4, skin toxicity was observed. So, on comparing formulation 4 with formulation 2m, the skin toxicity profile of formulation 2m is definitely better as no toxicity was observed till day 12. Although skin toxicity is a hurdle in studies involving nude mice, in humans, localized exposure of DTX due to extravasation has not been found significantly irritant or vesicant (<1% cases) (9). So, this type of toxicity is less likely to be encountered in humans.
Given the high IC50 value in the in vitro tests, it was logical to increase the amount of DTX in the formulation. But in this study, the amount of DTX in the formulation was kept the same; instead, a faster release formulation was taken up for the further studies i.e., formulation 1m and 2m which were modifications of formulation 1 and 2 instead of formulation 3 and 4 (slow release formulation).
Formulation 1m was discontinued due to skin toxicity in nude mice. This toxicity could be caused by following two reasons: (1) ulceration caused by DTX and (2) tumor growth leading to tenderness of the skin which is prone to damage. However, no such toxicity has been reported on in vivo evaluation of stent formulation in small animals (10–12) with other drugs. As for formulation 2m and blank formulation, studies were continued for 12 days. By the end of this period, all the formulations and the control groups had no significant difference in the tumor volume and weight. Formulation 2m, however, exhibited relatively greater control on tumor growth which is explained in detail as follows. With mouse weight between 16–20 g in the 5-mg/kg IV group would receive 80–100 μg of DTX. Formulation 2m loading (94.82 ± 24.22 μg) is comparable to IV dose but is released over a longer duration. So, there was a modest benefit of localized formulation over IV formulation. In this study, toxicity of the formulation was not scrutinized extensively but it can be expected that localized formulation will have lower systemic toxicity as compared to IV formulation.
Drug-eluting non-vascular stents have remained elusive for various organs even after tremendous development in the vascular stents (13). This article deals with the development of the formulation and its evaluation for the advance stage esophageal cancer as a palliation option. Although results obtained from this study are not very encouraging, it raises several questions which need to be addressed in order to develop esophageal drug-eluting stent. First and foremost is what amount of DTX is required for formulation effectiveness? And second, which model should be used for the in vivo formulation evaluation? These two questions are interrelated since a suitable dose can only be selected on the basis of a robust in vivo disease model. Small animal (rodent) models are convenient but a relevant animal model is necessary in the future. Healthy rabbit and pig models (14,15) have been used for esophageal stent evaluation but complex relationship between stent reocclusion and stent migration (displacement from the place of insertion) warrants for development of a diseased model for stent evaluation. This aspect is complicated because a high dose of drug can reduce tumor size but potentially can aggravate stent migration. Another important issue raised by this study is the toxicity of DTX. As explained earlier, DTX has insignificant skin reactions on IV administration. Also, paclitaxel (same chemical class as that of DTX) has been evaluated in preclinical (16–19) as well as clinical studies (20–22) for stent development, but all the reports indicate manageable side effects like inflammation, fibrosis, etc. One way forward for DTX use would be to combine it with a drug having a complementary mode of action and non-overlapping toxicity; this will reduce the dose as well as increase efficacy. For example, capacitabine (prodrug of 5-fluorouracil) (23) and cisplatin (24) can be combined with DTX for improved safety and efficacy.
CONCLUSION
From the above investigation, it can be concluded that formulations show marginal activity at the loading levels studied in the OE-19 and OE-21 EC cell lines. It was also found that DTX was relatively non-toxic to the normal cell line, Het-1A compared to OE-19 and OE-21 cell line. In vivo, localized DTX formulation performed fairly as compared to IV dosing but the effect of IV formulation was not significantly different from the blank formulation. However, to improve the outcome of DTX formulation, combination with a drug causing apoptosis by an alternate mechanism and non-overlapping toxicity is recommended.
References
Rashid N, Elshaer M, Kosmin M, Riaz A. Current management of oesophageal cancer. Br J Med Pract. 2015;8:a804.
Katsanos K, Sabharwal T, Adam A. Stenting of the upper gastrointestinal tract: current status. Cardiovasc Intervent Radiol. 2010;33:690–705.
Sharma P, Kozarek R. Role of esophageal stents in benign and malignant diseases. Am J Gastroenterol. 2010;105:258–73.
Martinez JC, Puc MM, Quiros RM. Esophageal stenting in the setting of malignancy. ISRN Gastroenterol. 2011;2011:719575.
FDA. Labelling informaton- Taxotere (Docetaxel) Injection, Sanofi-Aventis U.S. LLC. 1996. http://www.accessdata.fda.gov/drugsatfda_docs/label/2007/020449s045lbl.pdf. Accessed 4 Sept 2015.
Shaikh M, Choudhury NR, Knott R, Garg S. Engineering stent based delivery system for oesophageal cancer using Docetaxel. Mol Pharmaceutics. 2015;12:2305–17.
Liu R, Wolinsky JB, Walpole J, Southard E, Chirieac LR, Grinstaff MW, et al. Prevention of local tumor recurrence following surgery using low-dose chemotherapeutic polymer films. Ann Surg Oncol. 2010;17:1203–13.
Bhalla KN. Microtubule-targeted anticancer agents and apoptosis. Oncogene. 2003;22:9075–86.
Barbee MS, Owonikoko TK, Harvey RD. Taxanes: vesicants, irritants, or just irritating? Ther Adv Med Oncol. 2014;6:16–20.
Machan L. Clinical experience and applications of drug-eluting stents in the noncoronary vasculature, bile duct and esophagus. Adv Drug Deliv Rev. 2006;58:447–62.
Moon S, Yang SG, Na K. An acetylated polysaccharide-PTFE membrane-covered stent for the delivery of gemcitabine for treatment of gastrointestinal cancer and related stenosis. Biomaterials. 2011;32:3603–10.
Lee JW, Yang SG, Na K. Gemcitabine-releasing polymeric films for covered self-expandable metallic stent in treatment of gastrointestinal cancer. Int J Pharm. 2012;427:276–83.
Shaikh M, Kichenadasse G, Choudhury NR, Butler R, Garg S. Non-vascular drug eluting stents as localized controlled drug delivery platform: preclinical and clinical experience. J Controlled Release. 2013;172:105–17.
Liu J, Wang Z, Wu K, Li J, Chen W, Shen Y, et al. Paclitaxel or 5-fluorouracil/esophageal stent combinations as a novel approach for the treatment of esophageal cancer. Biomaterials. 2015;53:592–9.
Wang Z, Liu J, Wu K, Shen Y, Mao A, Li J, et al. Nitinol stents loaded with a high dose of antitumor 5-fluorouracil or paclitaxel: esophageal tissue responses in a porcine model. Gastrointest Endosc. 2015;82:153–60.
Lee DK, Kim HS, Kim KS, Lee WJ, Kim HK, Won YH, et al. The effect on porcine bile duct of a metallic stent covered with a paclitaxel-incorporated membrane. Gastrointest Endosc. 2005;61:296–301.
Herrmann BW, Citardi MJ, Vogler G, Gardner L, Smith G, Javer AR, et al. A preliminary report on the effects of paclitaxel-impregnated stents on sheep nasal mucosa. Am J Rhinol. 2004;18:119–24.
Jang SI, Kim JH, Kim M, Yang S, Jo EA, Lee JW, et al. Porcine feasibility and safety study of a new paclitaxel-eluting biliary stent with a Pluronic-containing membrane. Endoscopy. 2012;44:825–31.
Jeon SR, Eun SH, Shim CS, Ryu CB, Kim JO, Cho JY, et al. Effect of drug-eluting metal stents in benign esophageal stricture: an in vivo animal study. Endoscopy. 2009;41:449–56.
Suk KT, Kim JW, Kim HS, Baik SK, Oh SJ, Lee SJ, et al. Human application of a metallic stent covered with a paclitaxel-incorporated membrane for malignant biliary obstruction: multicenter pilot study. Gastrointest Endosc. 2007;66:798–803.
Song TJ, Lee SS, Yun SC, Park do H, Seo DW, Lee SK, et al. Paclitaxel-eluting covered metal stents versus covered metal stents for distal malignant biliary obstruction: a prospective comparative pilot study. Gastrointest Endosc. 2011;73:727–33.
Shah PL, Slebos DJ, Cardoso PF, Cetti E, Voelker K, Levine B, et al. Bronchoscopic lung-volume reduction with Exhale airway stents for emphysema (EASE trial): randomised, sham-controlled, multicentre trial. Lancet. 2011;378:997–1005.
Park YH, Ryoo BY, Choi SJ, Kim HT. A phase II study of capecitabine and docetaxel combination chemotherapy in patients with advanced gastric cancer. Br J Cancer. 2004;90:1329–33.
Ridwelski K, Gebauer T, Fahlke J, Kroning H, Kettner E, Meyer F, et al. Combination chemotherapy with docetaxel and cisplatin for locally advanced and metastatic gastric cancer. Ann Oncol. 2001;12:47–51.
Acknowledgments
The authors would like to acknowledge Professor Wayne Phillips (Surgical Oncology Research Laboratory, Peter MacCallum Cancer Centre, Melbourne) for providing OE-19, OE-21, and Het-1A cell lines.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This study was conducted in the School of Pharmacy, Shandong University as per the approval granted by the Animal Ethics Committee of Shandong University (ECAESDUSM).
Rights and permissions
About this article

Cite this article
Shaikh, M., Zhang, H., Wang, H. et al. In Vitro and In Vivo Assessment of Docetaxel Formulation Developed for Esophageal Stents. AAPS PharmSciTech 18, 130–137 (2017). https://doi.org/10.1208/s12249-016-0501-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-016-0501-7