

Abstract
We have prepared iron doped TiO2 samples by the impregnation of TiO2 (Degussa—P25) with Fe(III) nitrate as an iron precursor. Obtained materials were then characterized by structural x-ray diffraction (XRD) and optical (Uv-vis, Raman) techniques. Later, we deposited very small gold nanoparticles (AuNPs; <5 nm) onto as-prepared supports by a colloidal deposition method, in which lysine was used as a capping agent and a short sonication time was applied to facilitate the dispersion and deposition of AuNPs. The presence of Fe+3 in the TiO2 structure was confirmed by an increase in the number of oxygen defect sites that react with O2 to form reactive oxygen species. Transmission electron microscopy (TEM) was used to determine the size of the gold particles before and after deposition. The AuNPs deposited on the FeTiO2 exhibited good thermal stability against sintering, even at 500 °C for 4 h.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Since Haruta's initial report [1] on the exceptional catalyst properties in the CO oxidation reaction of supported gold nanoparticles (AuNPs), when their sizes are in the range of several nanometers, research efforts have been directed towards enhancing the activity of the catalyst by developing many methods of catalyst preparation and pretreatment conditions. The effect of gold particle size is considered to be an important parameter controlling catalyst activity. Both Haruta [2] and Goodman [3] reported that, for CO oxidation, the catalytic activity increases as the mean particle size decreases, and the optimum particle size is between 2 and 4 nm. However, the nature of support on which the small AuNPs are dispersed also plays a crucial role in the CO oxidation process. It is widely believed that oxygen absorption occurs at the metal–support interface, possibly in oxygen vacancies that should be present on semiconductor materials (e.g. TiO 2 and ZnO). Consequently, different activities observed for different catalysts were attributed to the different structures of the materials used as supports. Therefore, besides the need to have a good control of gold particle size, preparation of a specific catalyst support should also be considered.
Au/TiO 2 is one of the most active gold based catalysts for CO oxidation. The Au/TiO 2 interface and its role in CO oxidation have been studied in detail by numerous authors. Both bulk Au and TiO 2 absorb CO and O 2 weakly at ambient conditions, and some synergism between Au and TiO 2 seems to be the key to activation. Baikers [4] proposed that oxygen absorption takes place on TiO 2 only in the presence of vacancy defects:
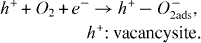
At the interface Au/TiO 2, the reactive oxygen species can move from TiO 2 to Au, the spillover rate of reactive oxygen species from TiO 2 to Au depends on the mean size of the AuNPs and the oxygen vacancy density in the support structure. Hence, the possibility of modifying a support to increase the availability of reactive oxygen species is of general interest in the design of active gold based catalysts.
In recent years, the technique for modifying the electrical properties of TiO 2 by metal ion doping has been widely studied. Among various dopants, substitution of metal iron (III) ions in the TiO 2 lattice is most favored due to the similar size of Fe +3 and Ti +4. However, most of these papers deal with photocatalytic application [5–7]. The recent report of Corma [8] demonstrated that the iron doped TiO 2 increases significantly the oxidation catalyst activity in comparison with the most active reported Au/TiO 2 catalyst. In the present work, we have studied the preparation and structural characterization of a nano-gold catalyst supported on iron doped TiO 2. Iron doping was carried out by a conventional impregnation method. The effect of the iron dopant was to insert oxygen vacancies in TiO 2 structure via substitution of Ti +4 by Fe +3. This effect was observed by XRD, Uv-vis and Raman spectroscopy techniques. The colloidal deposition process that was used in this study provides good control over particle size, in which HAuCl 4 was reduced instantly by NaBH 4 in the presence of lysine and catalyst support under a short period of ultrasound irradiation. TEM analysis was used to identify the particle dimension of gold and estimate the stability of AuNPs on catalyst support against sintering after thermal treatment.
2. Experimental
2.1. Catalyst preparation
All chemicals used were of p.a. purity and were used without further purification. De-ionized water was used for aqueous solution.
Iron doped TiO 2 powders were prepared by the impregnation of TiO 2 (Degussa P25) with aqueous solution containing the different amounts of Fe(NO 3)3·6H 2 O (Aldrich), then the slurry was stirred at 60 °C until all the liquid had evaporated. The obtained solids were dried at 100 °C overnight and then calcined at 600 °C in a static oven for 5 h. Four samples were prepared with nominal concentrations alternately: 0.5%, 1% and 2% (0.5% FeTiO 2, 1% FeTiO 2 and 2% FeTiO 2). For comparison, a sample of TiO 2 P25 (0%) was also calcined at 600 °C for 5 h.
For the gold colloidal deposition process, we used a one-stage method, in which gold colloids were produced in situ in the presence of catalyst support. In a typical preparation, 5 ml HAuCl 4 0.01 N (Merck) and 10 ml lysine 0.01 N (Merck) were diluted in a volume of water, and 0.5 g Fe–TiO 2 was added to the solution. After stirring for 5 min, the pH of the suspension was adjusted to 8–9 with 0.02 N of NaOH solution (China). The suspension was subjected to sonication (Vibracell 500 Watt Ultrasonic processor, 40%) for 60 s, and during the sonication freshly prepared NaBH 4 0.1 N (Merck) was injected instantly. The suspension immediately turned dark in color (orange–brown), indicating the formation of very small AuNPs. The mixture was then continuously stirred for 30 min. The resulting solid (dark powders) was washed for several times with 2 liters of de-ionized water using a centrifuge in order to remove all of Cl − species. Finally, the solid was dried under vacuum in the dessicators where P 2 O 5 (Aldrich 97%) as a drying agent was scattered on the bottom.
2.2. Catalyst characterization
X-ray diffraction: XRD patterns were taken at room temperature using a Bruker counter diffractometer model D8 ANCE (Cu-Kα radiation) at an angle of 20° to 80° to determine the crystal phase composition of the modified TiO 2 samples.
Uv-vis spectroscopy: Uv-vis spectra of the prepared supports were collected with a CARY 100 instrument (Variance) in the wavelength range from 200 to 800 nm.
Raman spectroscopy: Raman experiments were performed on iron doped TiO 2 powders after being finely deposited on a glass slide substrate. A coherent laser with λ=514.5 nm served as the excitation source and the scattered light was analyzed with a LABRAM 300—JOBINYVON Raman spectrometer.
Microscopy experiments: Transmission electron microscopy (TEM) analysis was performed on gold loaded samples by using a JEM 1010—JEOL microscope. The particle size and the particle size distribution were determined from the TEMs by measuring the size of 200 typical particles.
3. Results and discussions
3.1. Iron doped titanium oxide characterization
3.1.1. X-ray diffraction.
The presence of different amounts of iron in TiO 2 leads firstly to a change in appearance of the powder sample color from white to yellow (0.5%), to orange (1%) and to orange–brown (2%).
The XRD patterns of the nondoped and doped samples are displayed in figure 1, showing the presence of typical peaks attributed to two crystal phases—anatase (A) and rutil (R)—where the former is dominant. In these diffractograms, noncharacteristic peaks related to iron containing phases, like hematite (α Fe 2 O 3) or pseudo brookite (Fe 2 TiO 5), could be resolved. Diameters of the anatase crystal particles calculated using the Scherre diffraction formula are about 20.4 nm for all samples. Two suggestions can be proposed: (i) the Fe +3 may have been moved either into interstitial positions or substitutional sites of the TiO 2 crystal structure and (ii) the iron segregation could be produced in the doped samples but their crystal nature is too subtle to be detected by this instrument.

Figure 1 XRD pattern TiO 2 P25 and Fe doped TiO 2 samples, IA=anatase, IR=rutil.
The TiO 2 P25 structure seems to be preserved for all doped samples. However, the iron doping could change the anatase and rutil composition, as reported by Corna [8]. The dispersion of Fe +3 and TiO 2 leads to catalyze the transformation of anatase to rutil, which in principle can stabilize a larger number of defects than pure anatase [8]. The amount of rutil phase transformed (F R ) was calculated according to the method described elsewhere [9]: F R =1/( 1+0.79IA/IR), where F R is the mass fraction of rutil in the samples, and IA and IR the integrated (101) intensities of anatase and (110) of rutil, respectively. F R was respectively 0.17, 0.26, 0.26, 0.20 with an increase in iron content (0–2%). It can be concluded that the R/A ratio is higher than that of nondoped TiO 2, and the formation of rutil phase is accelerated by the presence of iron during thermal treatment but the relation between this acceleration effect and the nominal concentration of iron could not be found.
3.1.2. Uv-vis spectra.
Uv-vis absorption spectra of TiO 2 doped with various amounts of iron are shown in figure 2. A step increase in the absorption at ∼400 nm can be assigned to the intrinsic absorption band gap of TiO 2. It is obvious that the Uv-vis spectra of iron doped TiO 2 show a red shift (404–417 nm) relative to that of undoped TiO 2 with an increase in iron doping content. According to many reported papers [8], this shift could be related to a charge transfer of 3 d electrons from Fe +3 ions to the conduction band of TiO 2, consistent with the incorporation of Fe +3 into the TiO 2 matrix.

Figure 2 UV-vis absorption spectra of undoped and doped TiO 2 with various amounts of iron dopant.
3.1.3. Raman spectra.
Raman spectroscopy confirms the presence of the anatase and rutil phases of TiO 2 in all the samples, as shown in figure 3. The former is characterized by bands at 143, 396, 514 and 636 cm −1, corresponding to active modes expected for this tetragonal structure (3E g +2B 1g +1A 2g ) [11]. The band at 446 cm −1 is assigned to the E g mode of rutil [8]. It appears that no significant difference could be resolved from undoped TiO 2 and doped samples. But a careful inspection of the spectra in the frequency range of 1000–1600 cm −1 reveals the presence of a weak peak at 1315 cm −1, assigned to the second harmonic vibrations of hematite [8] that can be confirmed by the mixed oxide spectra (TiO 2+Fe 2 O 3, 1% Fe) (figures 4 and 5). However, the presence of this peak is insignificant for low iron dopant contents (0.5% FeTiO 2 and 1% FeTiO 2), and it begins to be observed up to 2% of iron content. That is consistent with the results of Justo's work [12], where iron doping by the impregnation method allows the formation of a homogenous structure with the proportion of iron lower than 2%.

Figure 3 Raman spectra of undoped and doped TiO 2 with various amounts of iron dopant.

Figure 4 Raman spectra of iron doped samples and mixed oxides (TiO 2+1% Fe 2 O 3) in the range of 1000–1600 cm −1.

Figure 5 Raman spectra of undoped TiO 2 and 1% FeTiO 2 in the presence of O 2.
In principle, the TiO 2 P25 can participate in the activation of O 2 by forming superoxide species, but their density is too low to be detected due to the small number of surface defects (oxygen vacancy density) [10]. In the iron doped TiO 2, the oxygen vacancy density increases as evidenced by the increased intensity of the band at 1123 cm −1, corresponding to the characteristic band of η1-superoxide species [8]. The incorporation of Fe +3 ions in the TiO 2 matrix creates the oxygen vacancies, which in the presence of O 2 provides the catalyst with highly reactive surface oxygen species that activate the CO oxidation. Corna [8] proposed that CO molecules are absorbed and activated on the gold clusters. The reactive superoxide species generated adjacent to Fe +3 lattices in the TiO 2 matrix will interact with absorbed CO to form CO 2.
3.2. Supported gold catalyst characterization
In the past ten years, the colloidal deposition method for the preparation of gold based catalysts has been intensively studied in order to replace the common methods, such as wetness impregnation, co-precipitation (CP) and deposition–precipitation (DP) [13–17]. It is well known that the conventional impregnation method fails to obtain good control on gold particle size and the contamination of chloride ion (Cl –) in catalyst is unavoidable. It seems that CP and DP are relatively more successful with high loading of gold and high activity of CO oxidation. However, these two methods do not completely avoid the contamination of Cl – ions that can accelerate the sintering of gold during heat treatment, as well as poison the active sites. Indeed, in the colloid method, gold exists in the metallic state and is always capped by ligand molecules. Cl – ions are chemically separated from the AuNPs and can be easily removed by washing. Moreover, the size of the AuNPs can be controlled before they are deposited on the support. Finally, the third reason to choose this method in this work is to eliminate the influence of the support on the formation of the AuNPs. This special feature allows the creation of identical AuNPs on undoped and iron doped support, independent of the presence of Fe +3 in the TiO 2 matrix. That helps us to evaluate the properties of the modified catalyst in comparison with usual one.
To obtain very small gold colloids, we studied the effects of composition proportion between the precursor (HAuCl 4), the ligand (lysine) and the reducing agent (NaBH 4), as well as the pH and the sonication duration on the size of gold colloids. Consistent with results reported by Zhong and his research group, who applied this method for a gold catalyst supported on Fe 2 O 3 [13], we found that the pH values before (pHi) as well as after (pHf) adding NaBH 4 are the key factors to achieve control over the gold particle size. In our study, we used the pHi value in place of pHf, as used by Zhong. In fact, there is a relation between these two values: the value of pHi determines that of pHf. The optimum value of pHi is in the range of 6.5–9. At lower pH (pH <6), the aggregation opportunities of gold colloids increases, when the pH is too high, the reduction in HAuCl 4 takes place before using the strong reducing agent like NaBH 4, which helps effectively to produce the fast nucleation to form tiny particles.
However, the pH influences the gold uptake efficiency on the support. With TiO 2 (isoelectric point IEP=6), at pH=8–9, the gold loading content is lower than that expected (control effect of pH and IEP on gold loading efficiency will be discussed elsewhere). This value can be improved by using ultrasound irradiation during the deposition process. The acoustic cavitations with high speed jets and waves with a scale of several hundreds of meters per second can effectively push the particle to hit the catalyst support [13]. Moreover, ultrasonic irradiation has a strong dispersing impact on the aggregation state of AuNPs. Consequently, only isolated spherical AuNPs are formed. Once small AuNPs are formed through sonication, they install rapidly on the titanium support. As a result, the one-stage method allows better control over the particle size of the catalyst compared with the two-stage one, in which the colloidal solution is stabilized by magnetic stirring for 30 min before deposition on the support. That can be confirmed by Uv-vis analysis on the instant gold sol after 30 s sonication and stabilized sol, as described earlier (figure 6). The red shift of absorption spectra of stabilized sol is attributed to the formation of larger AuNPs. However, excess sonication (more than 2 min) could cause secondary aggregation of the gold colloids. Probably the localized high pressure and temperature produced during cavitations are not beneficial in stabilizing the AuNPs because they are less stable thermodynamically.

Figure 6 Uv-vis spectra of gold sol: (a) 30 s sonication (b) 30 min magnetic stirring.
TEM analysis of stabilized sol, as prepared under the mentioned conditions without the presence of a support, is displayed in figure 7. The statistics of particle size and size distribution are determined by the Image J tool. a(%) is the amount (%) of particles whose sizes are in the range of indicated size value and q(%) is the amount (%) of particles whose sizes are smaller than the indicated size value. The TEM image indicates that all of the AuNPs were sphere-shaped, and separately dispersed in solution. 90% of them were less than 4 nm and the mean diameter was 3±1 nm. The size and dimensions of as synthesized AuNPs could be favorable for the preparation of highly active oxidation catalysts.

Figure 7 TEM analysis of stabilized gold sol without support presence.
TEM results from gold catalysts supported on TiO2 and 1%Fe TiO 2 before thermal treatment are presented in figure 8(a). The deposited AuNPs are quite uniform in size in both samples, with >90% of them smaller than 4 nm. The mean sizes of these two samples are similar and smaller than those of stabilized sol, which again shows the advantage of a one-stage method compared with a two-stage one. Additionally, in the same preparation conditions (with the same pH adjustment), the particle dimensions of the gold was independent of the nature of the support. The presence of Fe +3 in the TiO 2 structure does not influence the formation of the AuNPs—they are so created in solution before being installed on the support surface. This feature is quite different to others methods.

Figure 8 TEM analysis of gold supported on TiO 2 and 1% FeTiO 2 before (a) and after (b) thermal treatment.
The thermal stability of the supported gold catalyst was evaluated by TEM analysis after thermal treatment (calcinations at 500 °C for 4 h) (figure 8(b)). It is obvious that the change in particle size of (Au/1% FeTiO 2) is smaller than that of the usual one (Au/TiO 2). The particle aggregation was delayed when AuNPs were formed on an iron doped titantia surface. In severe conditions of thermal exposure (500 °C for 4 h), >90% of particles still remained smaller than 4 nm in diameter, while that was lower than 80% for another case. The increase in stability of the AuNPs could be attributed to the change in the electrical properties on the surface of the support or the change in the TiO 2 structure. The reduction in Cl – ion absorption on the surface by the presence of Fe +3, as Hung W C et al reported in a recent paper [18], could also prevent the sintering of AuNPs under heat treatment.
4. Conclusion
Iron doped TiO 2 with different iron contents prepared by the impregnation method were characterized by Uv-vis, x-ray and Raman spectra. Fe +3 ions were partly incorporated into the TiO 2 matrix by replacing the Ti +4 ions. The iron segregation becomes significant in the doped sample of higher than 1% Fe. The increase in reactive oxygen species density produced by introducing iron into the TiO 2 matrix was confirmed. The one-stage colloidal deposition proved to be a good method that allows one to obtain >90% of the AuNPs of 2–4 nm in diameter. The particle size of the gold is independent of the presence of iron in the support, but iron presence can prevent the sintering of AuNPs, even at 500 °C for 4 h.
Acknowledgment
The authors are grateful for the support from the Laboratory for Nanotechnology (LNT), Vietnam National University, Ho Chi Minh City.