
Abstract
Complete genomes of xenobiotic-degrading microorganisms provide valuable resources for researchers to understand molecular mechanisms involved in bioremediation. Despite the well-known ability of Sphingomonas paucimobilis to degrade persistent xenobiotic compounds, a complete genome sequencing is lacking for this organism. In line with this, we report the first complete genome sequence of Sphingomonas paucimobilis (strain AIMST S2), an organophosphate and hydrocarbon-degrading bacterium isolated from oil-polluted soil at Kedah, Malaysia. The genome was derived from a hybrid assembly of short and long reads generated by Illumina HiSeq and MinION, respectively. The assembly resulted in a single contig of 4,005,505 bases which consisted of 3,612 CDS and 56 tRNAs. An array of genes involved in xenobiotic degradation and plant-growth promoters were identified, suggesting its’ potential role as an effective microorganism in bioremediation and agriculture. Having reported the first complete genome of the species, this study will serve as a stepping stone for comparative genome analysis of Sphingomonas strains and other xenobiotic-degrading microorganisms as well as gene expression studies in organophosphate biodegradation.
Measurement(s) | genome • sequence_assembly |
Technology Type(s) | DNA sequencing • genome assembly |
Sample Characteristic - Organism | Sphingomonas paucimobilis |
Sample Characteristic - Environment | oil contaminated soil |
Sample Characteristic - Location | Malaysia |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.9971711
Similar content being viewed by others

Background and Summary
Sphingomonas spp. are Gram-negative, oxidase positive and non-fermentative rods1. One of the best known species of the genus is Sphingomonas paucimobilis as it was originally said to be the only species described in human infection1,2. It is a non-spore forming strictly aerobic, yellow-pigmented bacteria that can survive in low nutrient environment1,3. S. paucimobilis is naturally found in diverse environments such as soil and water and also has been shown to have a wide range of xenobiotic-biodegradative abilities4,5,6. Previous studies had shown its’ ability to degrade various types of hydrocarbons and pesticides, specifically chlorpyrifos7,8,9,10,11,12. It is also well recognized for its potential for biofilm formation13. Despite the potential role of this bacterium in bioremediation, there is a lack of complete genome in the public domain which will allow for the identification of genes involved in the biodegradation of chlorpyrifos, a widely used organophosphate.
General features of S. paucimobilis strain AIMST S2 are summarized in Table 1. S. paucimobilis strain AIMST S2 was first isolated in an oil-contaminated soil sample from Kedah, Malaysia. Following enrichment in LB broth, this strain was acclimatized in M9 minimal medium supplemented with diesel (max. 1% v/v) and chlorpyrifos (max. 100 mg/L) in increasing concentrations, as the sole carbon source. Genomic DNA extraction was performed according to the GeneJet Genomic DNA purification kit’s protocol using a log-phase culture grown in Luria broth. The concentration and quality of extracted DNA was determined using Nanodrop, Qubit dsDNA BR assay and a 1% (v/w) agarose gel. The genomic DNA was then subjected to sequencing via Illumina HiSeq. 2500 and Oxford Nanopore. DNA sequencing was performed with both Illumina and Nanopore technologies as they yield short (~150 bases) and long reads (~10,000 bases), respectively, a combination of which has shown to improve hybrid genome assembly quality by providing accurate, complete genomes without gaps14.
The complete genome sequence reported in this study will be useful for analysis of protein-coding gene families, identification of genomic islands, repeat regions, prophages, and structural rearrangements. Apart from that, the data from this study can be utilized for comparative genome analysis of strains belonging to the genus Sphingomonas and other xenobiotic-degrading microorganisms, as well as transcriptome studies of chlorpyrifos biodegradation.
An overview of the experimental design of the study is illustrated in Fig. 1 and a detailed account of the workflow is provided in the methodology.

Overview of the experimental design of study.
Methods
Bacterial growth and genomic DNA extraction
S. paucimobilis was cultivated in LB broth and incubated at 37 °C until it attained an absorbance of ~0.7 at 600 nm. The log-phase culture was centrifuged at 10,000 × g for 10 minutes and the cell pellet was subjected to genomic DNA extraction according to the GeneJet Genomic DNA purification kit’s protocol (Thermo Fisher Scientific, Waltham, MA, USA). The concentration and quality of extracted DNA was determined using Nanodrop ™ Lite spectrophotometer (Thermo Scientific, Wilmington, DE, USA), Qubit dsDNA BR assay (Thermo Scientific, Wilmington, DE, USA) and 1% (v/w) agarose gel electrophoresis. The genomic DNA was then subjected to sequencing via Illumina HiSeq. 2500 and MinION.
Illumina Sequencing
DNA was fragmented using Covaris to a targeted size of 350 bp and upon adapter ligation, a library containing fragments of 470 bp was generated. The library size was determined using Bioanalyzer high sensitivity DNA chip (Agilent, CA, USA). Library was prepared using NEBNext Ultra DNA Library Prep Kit for Illumina (NEB, MA, USA) and paired-end sequenced.
Oxford Nanopore MinION Sequencing
Approximately 500 ng genomic DNA was used to build a DNA library using a Rapid Sequencing Kit (SQK-RAD004) (ONT, Oxford, UK) as described by the manufacturer. MinKNOW software version 2.0 (ONT, Oxford, UK) was used to perform a quality check on the flow cell before the DNA library was loaded. Sequencing was performed on MK1B (MIN-101B) MinION platform with a FLO-MIN 106 R9.4 (SpotON) flow cell according to the manufacturers’ instructions. Raw sequence reads were basecalled real time using MinKNOW, producing Fastq format data.
Hybrid genome assembly
The FastQ format data obtained from Illumina and MinION sequencing was subjected to genome assembly using Unicycler version 0.4.3 with default parameters.
Genome annotation
The assembly was annotated with Prokka15. Genome-wide COG functional annotation was performed using eggNOG mapper with DIAMOND mapping mode, which is available in version 4.5.116,17. Following this, the amino acid sequences were subjected to KEGG analysis via KAAS for pathway mapping. Prophages and genomic islands were also identified using PHASTER18 and IslandViewer 419.
Data Records
Sequencing raw reads obtained from Illumina and Nanopore MinION runs have been deposited in the NCBI Sequence Read Archive under SRP185601 (accessible at https://identifiers.org/ncbi/insdc.sra:SRP185601)20. All predicted genes and their functional annotations are provided in GenBank (Accession number: NZ_CP035765)21. The circular genome assembly for S. paucimobilis has been deposited in NCBI Assembly under GCA_003314795.222, and the whole project is at BioProject under PRJNA478628 (https://identifiers.org/bioproject:PRJNA478628).
Technical Validation
FaQCs was used to obtain the sequencing statistics and Q scores of Illumina short-reads, while Pauvr was used to obtain the same for MinION sequencing (Table 2). Illumina sequencing yielded paired-end reads of ~150 bases with more than 98% reads possessing Phred scores (Q scores) above 20 (Fig. 2a), when quality screening was performed with FaQCs. MinION reads were also of high quality, as shown in Fig. 2b.

Phred analysis of Illumina and MinION reads for the Sphingomonas paucimobilis AIMST S2 strain genome. (a) Q scores for Illumina reads. (b) Q scores for MinION reads.
The hybrid genome assembly performed with the reads provided a complete, circular genome of S. paucimobilis, containing 4,005,505 bases, with an overall GC content of 65.73%. The sequencing coverage based on raw reads was 446.6×. A total of 3,612 coding sequences (CDS), 56 tRNAs, 1 tmRNA and 1 CRISPR array were identified. Three identical ribosomal operons were identified.
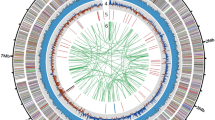
Figure 3 illustrates the circular genome of S. paucimobilis plotted using CGView23.

Circular map of S. paucimobilis. (a) Circular representation of genome with basic features including CDS and tRNA distributions. (b) Circular representation of genome based on COG classification.
Several levels of validation were performed to refine the hybrid assembly and check for completeness and the quality of genes predicted. Pilon refines the assembly using short reads during the final stage of assembly in Unicycler, by detecting and correcting single base differences, small and large indels or block substitution events. The present hybrid assembly was polished twice by Pilon with no changes in the assembly, suggesting an accurate assembly.
The completeness of the genomic data was further assessed according to Watson and Warr (2019)24. A DIAMOND blast against the UniProt TREMBL database showed that 99.1% of the genes predicted in the genome had more than 90% coverage to its top hit, suggesting good quality assembly and annotation was generated.
Among these, approximately 32 genes were shown to be involved in xenobiotic degradation (Table 3).
Interestingly, one of the key genes responsible for organophosphate biodegradation, glutathione S-transferase, gst was identified in the analysis. gst has previously been said to detoxify xenobiotics by catalyzing the nucleophilic conjugation of reduced tripeptide glutathione (GSH; γ-Glu-Cys-Gly) into hydrophobic and electrophilic substrates25,26.
Apart from genes involved in chlorpyrifos and other xenobiotic biodegradation, several genes related to plant-growth promoting factors were also identified in the genome. This includes several genes in auxin biosynthesis, alkaloid biosynthesis and nitrogen metabolism. Auxin plays a significant role in promoting stem elongation27,28, while alkaloid plays an important role in plants by preventing insects from eating them29. Genes involved in nitrogen metabolism like nitrate reductase, on the other hand, is responsible in reducing nitrate to nitrite for the production of protein in most crop plants, as nitrate is the predominant source of nitrogen in fertilized soils30,31,32.
Characterization of the complete genome of S. paucimobilis, identification of potential chlorpyrifos-degrading gene, gst and an array of genes coding for plant-growth promoting factors opens an avenue to more studies on bioremediation and its’ potential use as an effective microorganism in bioremediation and agriculture.
References
Ryan, M. P. & Adley, C. C. Sphingomonas paucimobilis: a persistent Gram-negative nosocomial infectious organism. J. Hosp. Infect. 75, 153–157 (2010).
Martínez, M. A. & Ovalle, A. Sphingomonas paucimobilis. Rev Chilena Infectol 30, 49–50 (2013).
Walayat, S., Malik, A., Hussain, N. & Lynch, T. Sphingomonas paucimobilis presenting as acute phlebitis: A case report. IDCases 11, 6–8 (2018).
Guo, C., Dang, Z., Wong, Y. & Tam, N. F. Biodegradation ability and dioxgenase genes of PAH-degrading Sphingomonas and Mycobacterium strains isolated from mangrove sediments. Int. Biodeter. Biodegr 64, 419–426 (2010).
Ayed, L., Mahdhi, A., Cheref, A. & Bakhrouf, A. Decolorization and degradation of azo dye Methyl Red by an isolated Sphingomonas paucimobilis: Biotoxicity and metabolites characterization. Desalination 274, 272–277 (2011).
Che Noraini, C. H., Morad, N., Norli, I., Teng, T. T. & Ogugbue, C. J. Methylene blue degradation by Sphingomonas paucimobilis under aerobic conditions. Water Air Soil Pollut 223, 5131–5142 (2012).
Li, X., He, J. & Li, S. Isolation of a chlorpyrifos-degrading bacterium, Sphingomonas sp. strain Dsp-2, and cloning of the mpd gene. Res. Microbiol. 158, 143–149 (2007).
Math, R. K. et al. Isolation of a novel gene encoding a 3,5,6-trichloro-2-pyridinol degrading enzyme from a cow rumen metagenomic library. Biodegradation 21, 565–573 (2010).
Singh, B. K., Walker, A., Morgan, J. A. W. & Wright, D. J. Effects of soil pH on the biodegradation of chlorpyrifos and isolation of a chlorpyrifos-degrading bacterium. Appl. Environ. Microbiol. 69, 5198–5206 (2003).
El-Helow, E. R., Badawy, M. E. I., Mabrouk, M. E. M., Mohamed, E. A. H. & El-Beshlawy, Y. M. Biodegradation of chlorpyrifos by a newly isolated Bacillus subtilis strain, Y242. Bioremediat. J. 17, 113–123 (2013).
Anwar, S., Liaquat, F., Khan, Q. M., Khalid, Z. M. & Iqbal, S. Biodegradation of chlorpyrifos and its hydrolysis product 3,5,6-trichloro-2-pyridinol by Bacillus pumilus strain C2A1. J. Hazard Mater. 168, 400–405 (2009).
Xu, G. et al. Biodegradation of chlorpyrifos and 3,5,6-trichloro-2-pyridinol by a newly isolated Paracoccus sp. strain TRP. Int. Biodeter. Biodegr. 62, 51–56 (2008).
Gulati, P. & Ghosh, M. Biofilm forming ability of Sphingomonas paucimobilis isolated from community drinking water systems on plumbing materials used in water distribution. J Water Health 15, 942–954 (2017).
Todd, S. M., Settlage, R. E., Lahmers, K. K. & Slade, D. J. Fusobacterium genomics using MinION and Illumina sequencing enables genome completion and correction. mSphere 3, e00269–18 (2018).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Huerta-Cepas, J. et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 44, D286–D293 (2016).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol Biol Evol 34, 2115–2122 (2017).
Arndt, D. et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44, W16–W21 (2016).
Bertelli, C. et al. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res 45, W30–W35 (2017).
NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP185601 (2019).
Ravintheran, S. K. et al. Sphingomonas paucimobilis strain AIMST S2 chromosome, complete genome. GenBank, https://identifiers.org/ncbi/insdc:CP035765 (2019).
NCBI Assembly, https://identifiers.org/ncbi/insdc.gca:GCA_003314795.2 (2018).
Grant, J. R. & Stothard, P. The CGView server: a comparative genomics tool for circular genomes. Nucleic Acids Res 36, W181–W184 (2008).
Watson, M. & Warr, A. Errors in long-read assemblies can critically affect protein prediction. Nat. Biotechnol. 37, 124 (2019).
Shen, M. et al. Identification of glutathione S-transferase (GST) genes from a dark septate endophytic fungus (Exophiala pisciphila) and their expression patterns under varied metals stress. PLoS One 10, e0123418 (2015).
Liu, Y.-J., Han, X.-M., Ren, L.-L., Yang, H.-L. & Zeng, Q.-Y. Functional divergence of the glutathione S-transferase supergene family in Physcomitrella patens reveals complex patterns of large gene family evolution in land plants. Plant Physiol. 161, 773–786 (2013).
Davies, P. J. In Plant Hormones: Biosynthesis, Signal Transduction, Action! (ed. Davies, P. J.) Ch. 1 The Plant Hormones: Their Nature, Occurrence, and Functions. (Springer Netherlands, 2010).
Santner, A., Calderon-Villalobos, L. I. A. & Estelle, M. Plant hormones are versatile chemical regulators of plant growth. Nat. Chem. Biol. 5, 301–307 (2009).
Steppuhn, A., Gase, K., Krock, B., Halitschke, R. & Baldwin, I. T. Nicotine’s defensive function in nature. PLoS Biol. 2, e217 (2004).
Crawford, N. M. & Guo, F.-Q. New insights into nitric oxide metabolism and regulatory functions. Trends Plant Sci 10, 195–200 (2005).
Ho, C.-H., Lin, S.-H., Hu, H.-C. & Tsay, Y.-F. CHL1 functions as a nitrate sensor in plants. Cell 138, 1184–1194 (2009).
Kaiser, W. M. & Huber, S. C. Post-translational regulation of nitrate reductase: mechanism, physiological relevance and environmental triggers. J Exp. Bot 52, 1981–1989 (2001).
Acknowledgements
We would like to acknowledge Malaysian Genomic Resource Centre (MGRC) for performing the Illumina sequencing.
Author information
Authors and Affiliations
Contributions
The project and pipeline were conceived and designed by H.R. and S.P. DNA extraction was performed by S.R. Sequencing was performed by S.S., S.R. and S.L. Computational resource was provided by A.M. Data analysis was performed by S.R., L.C., A.M., H.R. and S.P. The manuscript was written and revised by S.S., H.R., S.P., L.S.Y., R.M. and A.M. The final manuscript was approved by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Ravintheran, S.K., Sivaprakasam, S., Loke, S. et al. Complete genome sequence of Sphingomonas paucimobilis AIMST S2, a xenobiotic-degrading bacterium. Sci Data 6, 280 (2019). https://doi.org/10.1038/s41597-019-0289-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0289-x
