
Abstract
Using a new fossil-calibrated mitogenome-based approach, we identified macroevolutionary shifts in mitochondrial gene order among the freshwater mussels (Unionoidea). We show that the early Mesozoic divergence of the two Unionoidea clades, Margaritiferidae and Unionidae, was accompanied by a synchronous split in the gene arrangement in the female mitogenome (i.e., gene orders MF1 and UF1). Our results suggest that this macroevolutionary jump was completed within a relatively short time interval (95% HPD 201–226 Ma) that coincided with the Triassic–Jurassic mass extinction. Both gene orders have persisted within these clades for ~200 Ma. The monophyly of the so-called “problematic” Gonideinae taxa was supported by all the inferred phylogenies in this study using, for the first time, the M- and F-type mitogenomes either singly or combined. Within Gonideinae, two additional splits in the gene order (UF1 to UF2, UF2 to UF3) occurred in the Mesozoic and have persisted for ~150 and ~100 Ma, respectively. Finally, the mitogenomic results suggest ancient connections between freshwater basins of East Asia and Europe near the Cretaceous–Paleogene boundary, probably via a continuous paleo-river system or along the Tethys coastal line, which are well supported by at least three independent but almost synchronous divergence events.
Similar content being viewed by others


Introduction
The tempo, timing and mode of evolution have attracted considerable debate among evolutionary biologists. Here we use a new approach using mitogenome rearrangements to investigate changes at the geological time scale in the speciose and imperilled freshwater mussels.
In many taxonomic groups, the gene arrangement within mitogenomes is highly conserved, e.g., many vertebrate groups share the same gene order (Pereira 2000). Other faunal groups, such as the Bivalvia, exhibit a number of different mitochondrial gene arrangements (e.g., Yuan et al. 2012), which are the result of different mechanisms such as tandem duplication followed by gene loss (Boore 2000). Although local homoplastic arrangements have been identified in some invertebrate groups (e.g., Flook and Rowel 1995; Dowton and Austin 1999), complete gene orders generally remain unique and represent signatures with diagnostic value (Basso et al. 2017), providing a powerful signal for inferring ancient evolutionary relationships (Boore 2000).
Among freshwater mussels of the order Unionida, which spans about 900 species and represents the major bivalve radiation in the freshwater environment (Lopes-Lima et al. 2017a, 2018a), five mitogenome rearrangements have been described so far (Lopes-Lima et al. 2017b). Within the superfamily Unionoidea (Margaritiferidae+Unionidae), the mitochondria are furthermore unusual in that two highly divergent mtDNA molecules exist in males (Female or F- and Male or M-type) as a result of Doubly Uniparental Inheritance (DUI) (Zouros et al. 1994; Breton et al. 2009). This is in contrast to the vast majority of animal taxa, which inherit their mtDNA exclusively through the maternal lineage and thus exhibit only F-type mtDNA. In Unionoidea males, M-type mtDNA is restricted to the gonadal tissue inherited from the paternal lineage, and F-type mtDNA is present in all somatic tissues transmitted from the maternal lineage and also in female gonadal tissue (Breton et al. 2009; Froufe et al. 2016; Fonseca et al. 2016; Lopes-Lima et al. 2017b).
In recent decades, complete mitochondrial genome sequences have been published for a wide range of taxa, enabling reconstruction of shallow and deep phylogenies in both vertebrates and invertebrates (e.g., Jacobsen et al. 2014; Liu et al. 2016). However, the number of available mitogenomes for Unionida is low, particularly for M-type genomes (Froufe et al. 2016; Fonseca et al. 2016; Lopes-Lima et al. 2017b; Huang et al. 2019). A further shortcoming is that published mitogenomes are restricted to only a few higher Unionida taxa, with no mitogenomes being available for several families and subfamilies. In fact, of the six recognized Unionida families (Lopes-Lima et al. 2014), published mitogenomes are essentially restricted to the Unionoidea (Unionidae+Margaritiferidae) with a distribution predominantly within the Northern Hemisphere. While some studies have questioned the monophyly of the Unionoidea (e.g., Combosch et al. 2017; Whelan et al. 2011) the most comprehensive recent studies, using either full mitogenomes (Huang et al. 2019; Wu et al. 2019) or hundreds of nuclear loci (Pfeiffer et al. 2019) support its monophyletic status. Moreover, mitogenome-based Unionida phylogenies reconstructed to date have been based on either F- or M-type mitogenomes (Froufe et al. 2016; Fonseca et al. 2016; Lopes-Lima et al. 2017b). Although in these studies the highly divergent F- and M-type mitogenomes recovered identical phylogenies, concatenated phylogenetic analyses of M- and F-type datasets would be expected to recover a more robust phylogeny.
The Unionidae is the most species-rich Unionida family, comprising 620 species in several subfamilies and distributed widely (Lopes-Lima et al. 2017a). However, phylogenetic relationships within and between Unionidae subfamilies are still contentious and different phylogenies have been resolved with different analysed markers (e.g., Lopes-Lima et al. 2017a; Bolotov et al. 2017a).
One of the least studied Unionidae subfamilies, the Gonideinae, has a scattered distribution in the Northern Hemisphere (Lopes-Lima et al. 2017a). Species in this subfamily have suffered major declines, and half of the assessed Gonideinae species are currently listed as Near Threatened or Threatened (IUCN 2019). Moreover, 70% of recognized Gonideinae species have either never been assessed or are listed as Data Deficient by the IUCN Red List (IUCN 2019), indicating an urgent need for research into this family’s diversity, distribution and ecology.
Another outcome of the general lack of data on Gonideinae is their unresolved phylogeny. In fact, monophyly of this sub-family is disputed. The first molecular study to include the so-called “problematic” Gonideinae taxa (Graf 2002) only examined the type species, i.e., Gonidea angulata (Lea 1838). Subsequent studies included several additional Gonideinae taxa but the clade Gonideinae was never recovered as monophyletic (Graf and Cummings 2006; Whelan et al. 2011; Pfeiffer and Graf 2013). More recently, multi-marker and mitogenomic approaches have consistently recovered Gonideinae as monophyletic (Huang et al. 2013; Pfeiffer and Graf 2015; Fonseca et al. 2016; Froufe et al. 2016; Lopes-Lima et al. 2017a, 2017b). Bolotov et al. (2017a, 2017b) subsequently elevated one of the four Gonideinae tribes established by Lopes-Lima et al. (2017a), i.e., Pseudodontini, to the subfamily level (i.e., Pseudodontinae).
A good understanding of the evolutionary biogeography of the Gonideinae can be fundamental for reconstructing patterns of connections of freshwater systems through space and time on a global scale. Our knowledge in this respect is still far from complete. The first biogeographic scenarios developed using Unionida data (e.g., Starobogatov 1970; Banarescu 1991) proved highly inaccurate, as they were mostly descriptive and based solely on the (dis-)similarity between unionid faunas. Furthermore, these scenarios were generated at a time when unionid taxonomy was poorly resolved and included numerous paraphyletic higher-order taxa as well as nominal taxa, determined by shell shape rather than reliable indicators of true biological species (e.g., Bolotov et al. 2017a; Konopleva et al. 2017). Modern paleontology-based models seem to be much more reliable. Based on the fossil record from Vietnam, Schneider et al. (2013) developed the hypothesis of an independent development of Unionida faunas in the Yangtze and Mekong basins, at least during the entire Cenozoic. In addition, Van Damme et al. (2015) showed that the African Early Cretaceous Unionida are representatives of Asian/Eurasian taxa with the lack of Gondwanan elements, while the African Jurassic assemblages are distinctly related to those in Eurasia.
Recently, a first statistical biogeographic model for the Unionidae at the global level indicated that the Unionidae most likely originated in Southeast and East Asia in the Jurassic, with the earliest expansions into North America and Africa (since the Albian), following the colonization of Europe and India (Bolotov et al. 2017a). However, the Jurassic fossil record of western North America (for a review see Watters 2001) and Africa (Van Damme et al. 2015) indicate that these continents were colonized before the Cretaceous. Additionally, two species-rich monophyletic mussel radiations with an early Cenozoic or even pre-Cenozoic origin were discovered within the paleo-Mekong catchment (Bolotov et al. 2017a, 2017b). These findings revealed that the largest river systems (e.g., Mekong, Yangtze and Mississippi) may represent ancient evolutionary hotspots of freshwater mussels (Scholz and Glaubrecht 2004; Wesselingh 2007).
On the basis of the most comprehensive data set of mitogenomes sampled to date, including eight newly sequenced mitogenomes, this paper aims to improve our understanding of the higher-order phylogeny and classification of Unionidae by the following: (1) testing the monophyly of the poorly known Gonideinae subfamily using both full F- and M- mitogenomes and, for the first time, mitogenomes concatenated; (2) estimating macroevolutionary patterns in freshwater mussels of the Unionidae using, for the first time, a fossil-calibrated mitogenomic approach; (3) estimating the timing of major divergence events and comparing them to those of mitogenome rearrangements; and (4) developing an updated integrative approach to the systematics of Unionidae, on the basis of the mitogenomic results. This will allow the reconstruction of the potential origin and ancient radiations of the Unionidae and detect the most probable ancestral areas.
Methods
Sampling, DNA extractions, sequencing, assembly and annotation
One male specimen of Chamberlainia hainesiana, Microcondylaea bonellii, Pilsbryoconcha exilis and Monodontina vondembuschiana were dissected for sampling of gonadal (to recover M-type mtDNA) and mantle (to recover F-type mtDNA) tissues. DNA extractions followed Froufe et al. (2016). The complete M- and F-type mitogenome sequencing and assemblage followed Gan et al. (2014), while annotations were performed using MITOS (Bernt et al. 2013). The final limits of tRNA genes were rechecked with ARWEN (Laslett and Canbäck 2008). All F- and M- mitogenomes have been deposited in GenBank database under the accession numbers MK994770–MK994777 and were visualized using GenomeVx (Conant and Wolfe 2008).
DNA (NUC) and amino acid (AA) sequences of all mtDNA protein-coding genes (PCG), except ATP8 and the gender-specific open reading frames (M-ORF, H-ORF and F-ORF; Breton et al. 2011), were used in the phylogenetic analyses. The sequences of each gene were aligned using MAFFT software (version 7.304, Katoh and Standley 2013) and trimmed with GUIDANCE2 (Sela et al. 2015; see Froufe et al. (2016) for the parameters used).
The gene alignments were then concatenated, resulting in two alignments with the following length: 13449 aligned nucleotide positions and 3870 aligned amino acids positions + 1889 aligned nucleotide positions from the rRNA genes. The optimal partitioning scheme for each alignment was selected using PartitionFinder v. 2.1.1 software (Lanfear et al. 2016) under the greedy algorithm with proportional branch lengths across partitions. The best substitution models of DNA and protein evolution for each partition were selected under the BIC ranking method (Schwarz 1978). The codon positions of the protein-coding genes and each rRNA were defined as the initial data blocks for the partitioning schemes search.
An additional data set was also created, concatenating both F- and M-type gene alignments, with the following length: 26780 aligned nucleotide positions and 7661 aligned amino acid positions + 3797 aligned nucleotide positions from the rRNA genes. This alignment included 45 Unionida species plus Mytilus galloprovincialis as an outgroup (Table 1) using the same partitioning method and model selection as described above.
Phylogenetic analyses
All phylogenetic analyses were performed using both Maximum Likelihood (ML) and Bayesian Inference (BI) methods. ML analyses were performed using RAxML (v. 8.0.0, Stamatakis 2014) with 100 rapid bootstrap replicates and 20 ML searches. The BI was applied using MrBayes v. 3.2.7a (Ronquist et al. 2012) with two independent runs (107 generations with a sampling frequency of 1 tree for every 100 generations), each with four chains (3 hot and 1 cold). All runs reached convergence (average standard deviation of split frequencies below 0.01). The posterior distribution of trees was summarized in a 50% majority rule consensus tree (burn-in of 25%).
Divergence time estimates
The time-calibrated mitogenomic phylogeny was reconstructed in BEAST v. 1.8.4 based on two reliable fossil calibrations (Supplementary Table 1) using a lognormal relaxed clock algorithm with the Yule speciation process as the tree prior (Drummond et al. 2006, 2012; Drummond and Rambaut 2007). Calculations were performed at the San Diego Supercomputer Center through the CIPRES Science Gateway (Miller et al. 2010). The sample of M-type mitogenomes was used as outgroup. Similar settings to each gene partition as in the MrBayes analyses were specified but using a simplified evolutionary model (HKY; see Bolotov et al. 2017a for details). Five replicate BEAST searches were conducted, each with 5 × 107 generations and a tree sampling every 5000th generation. The log files were checked visually with Tracer v. 1.7 for an assessment of the convergence of the MCMC chains and the effective sample size of parameters (Rambaut et al. 2018). The chains in one run did not reach the convergence and were excluded, the other runs were compiled with LogCombiner v. 1.8.4 (Drummond et al. 2012) using an appropriate burn-in depending on the start of convergence of MCMC chains in each run. Most of ESS values were recorded as > 300, with a few ESS values > 100. The maximum clade credibility tree was obtained from the post-burn-in trees using TreeAnnotator v. 1.8.4 (Drummond et al. 2012).
Ancestral gene order and ancestral area reconstructions
TreeREx (Bernt et al. 2008) was used for inferring the most parsimonious putative ancestral gene orders and gene rearrangements along the obtained Unionidae F-haplotype phylogenetic sub-tree with the default settings (http://pacosy.informatik.uni-leipzig.de/185-0-TreeREx.html). Ancestral area reconstruction models were calculated for the Unionidae using three different approaches, i.e., Statistical Dispersal-Vicariance Analysis (S-DIVA), Dispersal-Extinction Cladogenesis (Lagrange configurator, DEC), and Statistical Dispersal-Extinction Cladogenesis (S-DEC) implemented in RASP v. 3.2 (Yu et al. 2015) following Bolotov et al. (2017a). Margaritiferidae were not used in this analysis due to the limited number of available mitogenomes. Four possible distribution areas of the in-group taxa were coded as follows: (A) Southeast Asia, (B) East Asia, (C) North America, and (D) Europe. From the input matrix, two geographically unreliable constrains (AC and AD) were excluded.
Results
Mitogenome characteristics and gene arrangements
All eight sequenced haplotypes include the 13 protein-coding genes (PCGs) typically found in metazoan mitochondrial genomes, the sex-specific ORF described for all Unionida mitogenomes with DUI system (Breton et al. 2009, 2011) and 22 transfer RNA (tRNA) and two ribosomal RNA (rRNA) genes (Fig. 1). As expected, the length of the four newly sequenced M-type mitogenomes is larger than the corresponding F-type (Breton et al. 2009), ranging from 16,267 bp in P. exilis to 17,465 bp in C. hainesiana, while the F-type ranged from 16,020 bp in M. bonellii to 16,746 bp in C. hainesiana (Table 2). The A+T content, and GC and AT skews are similar in all sequenced species in both F and M mtDNA types, averaging around 60%, 37 (+) and −0.23 (+), respectively (Table 2).

Gene maps of the F- and M-type mitochondrial genomes of Chamberlainia hainesiana, Microcondylaea bonellii, Pilsbryoconcha exilis and Monodontina vondembuschiana. Genes positioned inside the circle are encoded on the heavy strand, and genes outside the circle are encoded on the light strand. Color codes: Small and large ribosomal RNAs (red), transfer RNAs (purple), FORF F-specific open reading frame (yellow), MORF M-specific open reading frame (yellow), PCGs genes (green)
The gene arrangements of Microcondylaea bonellii, P. exilis and Monodontina vondembuschiana are identical to all Gonideinae mitogenomes available on GenBank (2018), named UF2 (Lopes-Lima et al. 2017b). However, C. hainesiana has a new and distinct gene arrangement, here named UF3 (Fig. 2).

Diagrams of the four distinct gene orders known in Unionidae to date. In the F-type, three gene orders are depicted: UF1, UF2 and UF3. In the male M-type lineage, the only Unionidae gene arrangement is shown: M-type 1 (UM1). Blue boxes highlight gene rearrangement region from UF1 to UF2 (Box A) and from UF2 to UF3 (Box B). Small and large ribosomal RNAs and transfer RNAs are depicted by one letter of the amino acid code; Arrow colour codes, follow Fig. 1
Phylogenetic analyses
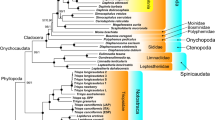
All the phylogenies inferred in this study that are based on M and F mitogenomes alone (i.e., not combined) support the monophyly of Gonideinae (Fig. 3). Moreover, the four tribes Chamberlainiini, Gonideini, Lamprotulini and Pilsbryoconchini, are also monophyletic in both M- and F-type trees (Fig. 3). The same results were obtained when using for the first time the M and F mitogenomes combined, despite the lower number of species (Fig. 4). The only unsupported result on the topology is seen in the relationship among the tribes Gonideini, Pilsbryoconchini and Lamprotulini in the ML AA data set (Fig. 4).

Phylogenetic (BI-NUC) tree of Unionida estimated from 14 concatenated individual mtDNA gene sequences (12 protein-coding and 2 rRNA genes). Values for branch support are represented in the following order: (1) Bayesian posterior probabilities (PP) for BI-NUC tree, (2) Bayesian PP for BI-AA tree, (3) ML bootstrap support (BS) values for ML-NUC and (4) ML BS values for ML-AA tree. Maximum support values (PP = 1, BS = 100) are represented by asterisks. Gonideinae subfamily and tribes are highlighted. For details see text. GenBank codes in Table 1

Phylogenetic (BI-NUC) tree of Unionida estimated from 28 concatenated individual mtDNA gene sequences (24 protein-coding and 4 rRNA genes) of the first combined Female+Male concatenated data set. Maximum branch support values (BI-NUC/BI-AA PP = 1; ML-NUC/ML-AA BS = 100) are represented by asterisks, while # represents the only non-supported branch by ML-AA tree. Gonideinae subfamily and tribes are highlighted. GenBank codes in Table 1
Ancestral gene order and ancestral area reconstructions
The TreeREx analysis indicated that the evolution of gene orders in the Unionidae F-type mtDNA is characterized by two independent events of tandem duplication and random loss (TDRL), with every ancestral gene order showing the highest consistency scores. The analysis suggests that the ancestral gene order for Unionidae F mitogenome is UF1, which is also found in the contemporary species of the subfamilies Ambleminae and Unioninae (Fig. 5). The fossil-calibrated mitogenomic analysis placed the split between the UF1 and MF1 gene orders in the Late Triassic (mean age = 208 Ma, 95% high posterior density (HPD) 201–226 Ma) (Fig. 6).

Unionidae F-haplotype phylogenetic sub-tree (BI-NUC) used to infer the most parsimonious putative ancestral gene orders and gene rearrangements, mapped as MF1, UF1, UF2 and UF3 (see text for details). Margaritiferidae and all subfamily nodes were collapsed for visual purposes

Time-calibrated mitogenomic phylogeny, an example of three-level classification scheme (subfamilies, tribes and subtribes) and evolution of the mitochondrial gene order in the Unionoidea. Fossil-calibrated ultrametric chronogram of the Unionoidea calculated under a lognormal relaxed clock model and a Yule process speciation implemented in BEAST and obtained for the complete mitogenome data set. The outgroup sample is not shown. Bars indicate 95% confidence intervals of the estimated divergence times between lineages (Ma). Black numbers near nodes are mean ages (Ma). Color labels indicate the mitochondrial gene order (MF1, UF1, UF2, and UF3). Red asterisks indicate fossil calibrations (Supplementary Table 1). Stratigraphic chart according to the International Commission on Stratigraphy, 2015
The ancestral gene order of the Gonideinae species represented in our study is UF2, which results from a TDRL event of an mtDNA segment involving nad3, trnH, trnA, trnS2, trnS1, trnE, nad2, and trnM (Fig. 2 Box A). In UF2, the genes trnH, trnS1, nad2 and trnM pertain to the original segment, while the remaining genes–nad3, trnA, trnS2, and trnE–are present in the duplicated segment (Fig. 2 Box A). The fossil-calibrated model developed suggests that the UF1 and UF2 gene orders split near the Jurassic–Cretaceous boundary (mean age = 149 Ma, 95% HPD 138–162 Ma) (Fig. 6).
Finally, the UF3 gene order also arises after a TDRL event within Gonideinae (Fig. 2 Box B). It involved an mtDNA segment containing twelve genes: trnQ, trnC, trnI, trnV, trnL2, nad1, trnG, nad6, nad4, nad4l, atp8 and trnD. In UF3, the genes trnC, trnI, trnV, trnG, nad6, atp8 and trnD are retained in the original segment, whilst genes trnQ, trnL2, nad1, nad4 and nad4l were not lost in the duplicated one (Fig. 2 Box B). The fossil-calibrated model placed the split between the UF2 and UF3 gene orders in the Cretaceous near the Albian–Cenomanian boundary (mean age = 102 Ma, 95% HPD 77–124 Ma) (Fig. 6).
The combined ancestral area reconstruction model suggests that the Most Recent Common Ancestor (MRCA) of the crown group of the Ambleminae+(Gonideinae+Unioninae) clade used to be widely distributed across the supercontinent of Laurasia (probability 100%) (Fig. 7). The earliest split was between the Laurentian (Ambleminae) and Eurasian (Gonideinae+Unioninae) taxa. This vicariance event is placed in the Late Jurassic (mean age = 159 Ma, 95% HPD 155–170 Ma). Early diversification of the Gonideinae+Unioninae clade is placed within East Asia (probability 100%; Fig. 7). The origin of the MRCA of this large clade (mean age = 149 Ma, 95% HPD 138–162 Ma) and subsequent splitting into two subclades (mean ages of crown groups = 137 and 106 Ma and 95% HPD 123–152 and 90–124 Ma for Gonideinae and Unioninae, respectively) most likely resulted from an intra-area radiation (probability 100% in each case) during the early Cretaceous. The Yangtze and Mekong unionid faunas have likely been separated since the Albian (mean ages = 95–102 Ma, 95% HPD 77–124 Ma) (Fig. 7).

Historical biogeography of the Unionidae. This combined scenario has been inferred from three different statistical modeling approaches (S-DIVA, DEC and S-DEC) based on the time-calibrated mitogenomic phylogeny (Fig. 6). Pie charts near nodes indicate probabilities of certain ancestral areas. Color circles on the tip nodes indicate the range of each species. Color labels indicate the mitochondrial gene order (UF1, UF2, and UF3)
Discussion
Phylogenetic patterns
The new mitogenomic results presented here place the Pilsbryoconchina subtribe (previously under the Pseudodontinae as described by Bolotov et al. 2017a) as a subclade within the monophyletic Gonideinae in both the M- and F-type phylogenies. Our results are thus in agreement with the phylogeny recovered by Lopes-Lima et al. (2017a), which is also supported by morphological data. However, the recovered results contradict that of Bolotov et al. (2017a, 2017b), which suggested elevation of the Pseudodontini to the subfamily level.
Our results further indicate that the number of recognized subfamilies within the Unionidae is most likely lower than has been suggested by recent phylogenetic studies (Lopes-Lima et al. 2017a, 2017b; Bolotov et al. 2017a, 2017b). The mitogenomic results fully support three large subfamily-level clades: Ambleminae, Gonideinae and Unioninae. It is important to note that our analyses did not include members of the Parreysiinae and Rectidentinae. Nor did it include sequences of Modellnaia siamensis, the only species of the monotypic Modellnaiinae, which is characterized by a number of morphological and anatomical autapomorphies suggesting its separation within the Unionidae as a “phylogenetic relic” (Brandt 1974; Heard and Hanning 1978). Future studies including full mitogenomes of several taxa from Parreysiinae, Rectidentinae and Modellnaiinae are needed to fully resolve the higher-level phylogeny of the global Unionidae.
Our results highlight that resolving the systematics of a large, species-rich clade such as the Unionidae is a complex task. Previous taxonomic schemes for the Unionidae included only two levels of family-group names, i.e., subfamilies and tribes (reviews: Lopes-Lima et al. 2017a; Bolotov et al. 2017a, 2017b). However, our whole mitogenome analyses reveal that despite the limited number of taxa included, the Unionidae classification scheme could be better explained by including three levels of family-group names (i.e., subfamilies, tribes and subtribes) to accurately reflect the presence of several levels of highly divergent clades within this family (Fig. 6). Subfamilies represent the largest clades that are fully supported by the mitogenomic approach (Fig. 7); some of which may be characterized by unique morphological synapomorphies, although several subfamilies have been supported by molecular data only (e.g., Lopes-Lima et al. 2017a).
The most recent nuclear-based Unionoida phylogeny (using hundreds of nuclear protein-coding loci; Pfeiffer et al. 2019) shows strong similarity to our own findings in regard to the relationships of both families and subfamilies. Moreover, mitogenome data currently available suggest that the Unionidae comprise seven (Lopes-Lima et al. 2017a) or eight (Bolotov et al. 2017a) subfamily clades. Of these, the Gonideinae (encompassing Pseudodontinae), Unioninae (encompassing the Anodontinae) and Ambleminae were well supported in the mitogenomic results obtained herein, whilst the validity and placement of the Parreysiinae, Rectidentinae and Modellnaiinae clades are yet to be confirmed by mitogenomic analyses.
The largest monophyletic clades, within each subfamily, exhibiting significant morphological synapomorphies and fully supported by the present mitogenomic results, are herein considered as tribes. Therefore, using these criteria, the Gonideinae comprise two tribes, i.e., Gonideini (trapezoidal to rectangular shells with none or only vestigial hinge teeth and tetragenous brooding type) and Chamberlainiini (round to oval shells, with a well-developed hinge structure and ectobranchous brooding type).
The subtribes represent smaller but distant clades within the tribes, comprising several genera or even a single highly divergent genus that usually does not reveal any unique synapomorphies but can be distinguished on the basis of molecular characters. Based on data available to date, including the present results, the Gonideini comprise at least five subtribes, i.e., Chamberlainiina, Gonideina, Lamprotulina, Pilsbryoconchina and Pseudodontina (Lopes-Lima et al. 2017a; Bolotov et al. 2017a, 2017b).
Macroevolutionary patterns of the Unionidae
The new mitogenomic analysis presented herein supports the hypothesis of an ancient Mesozoic origin and diversification of the Unionoidea (Taylor 1988; Ma 1996; Van Damme et al. 2015; Bolotov et al. 2016; Araujo et al. 2017; Bolotov et al. 2017a, 2017b). The new results indicate that the Late Triassic split between the Margaritiferidae and Unionidae coincided approximately with the Triassic–Jurassic extinction that was one of the largest mass extinction events in the Phanerozoic (Watters 2001; Hesselbo et al. 2002; Bogan and Weaver 2012; Percival et al. 2017; Smithwick and Stubbs 2018). The divergence event between the two families was associated with TDRL event leading to formation of the two stable mitochondrial gene orders, i.e., MF1 and UF1, which have persisted without changes for ~200 Ma. However, there were at least two additional Mesozoic splits in the mitochondrial gene order (i.e., UF1 vs. UF2 and UF2 vs. UF3) within the Unionidae, with UF2 and UF3 being restricted to a single subfamily, the Gonideinae. The first split coincided with the origin of this subfamily but the UF3 is a third, new and distinct gene arrangement derived from UF2 present in a single species, Chamberlainia hainesiana. These two mitochondrial gene orders have also persisted for long-term periods of ~150 and ~100 Ma for UF2 and UF3, respectively.
At least two splits in the mitochondrial gene order were associated with the origin of the MRCAs of large and diverse clades of family (Unionidae vs. Margaritiferidae) or subfamily (Unioninae vs. Gonideinae) levels. With respect to this evidence, these TDRL events could be considered progressive evolutionary innovations because they lead to formation of stable gene orders that have persisted within widely distributed and diverse clades for ~150–200 Ma. As for the mitogenome gene order, our ancestral state analyses suggest UF1 (in the Unionidae) as the ancestral gene order, which is maintained in the subfamilies Ambleminae and Unioninae sensu lato (Fig. 6). Additionally, it indicates that the evolution of F-type mtDNA gene orders is characterized by two independent events of TDRL (Moritz et al. 1987; Boore 2000). One resulted in the evolution of UF2, present in the Gonideinae, and the other in UF3, within Gonideinae but restricted to Chamberlainia hainesiana. In contrast, all sequenced M-type Unionidae mitogenomes to date present the same gene order, i.e., UM1 (Lopes-Lima et al. 2017b) (Fig. 2). Possibly this could be explained by the higher natural selection pressure and/or due to the tight control of the DUI system on the paternal mitochondrial inheritance. In summary, our results reveal that each TDRL event was followed by the stable long-term persistence of a mitochondrial gene order through evolving lineages (or even a single lineage, although the Chamberlainia clade may actually be under-sampled) and corresponds to the first reliable mitogenomic evidence supporting the evolutionary stasis in molecular traits of freshwater bivalves. However, this should be further explored using an expanded data set of mitochondrial genomes that may facilitate the understanding of how evolutionary rates have shifted across multiple genetic loci and how that corresponds to ecologically relevant traits.
Diversification and Biogeography
Combining our new fossil-calibrated mitogenomic analyses with robust ancestral area reconstruction provides new insights into early diversification patterns and biogeography of the Unionidae. According to our results, the Ambleminae+(Gonideinae+Unioninae) clade originated in the late Jurassic, with their MRCA distributed across Laurentia and Eurasia of the supercontinent of Laurasia. The split between the Ambleminae and Gonideinae+Unioninae clades was likely associated with a vicariance event driven by plate tectonics, i.e., the formation of the early Jurassic Transcontinental Laurasian Seaway (Bjerrum et al. 2001). The Ambleminae is an entirely Laurentian subfamily, which diversified primarily through radiation within the Mississippi drainage basin from the Early Cretaceous (Bolotov et al. 2017a). In this context, a peculiar Unionidae fauna from the Late Jurassic of western North America (Watters 2001) appears to be ancestral lineages and stem groups of the Ambleminae+(Gonideinae+Unioninae) clade. The Gonideinae and the Unioninae (Unionini, Anodontini, Lanceolariini, and Lepidodesmini) (Fig. 6) originated in East Asia, most likely via intra-area radiation within the paleo-Yangtze River system during the Cretaceous (Fang et al. 2009; Wang et al. 2018). The Southeast Asian Gonideinae taxa (Mekong basin) were separated via several vicariance events in the Albian - Cenomanian, which may indicate the drainage rearrangement of paleo-river systems of the Indochina Peninsula and surrounding terrains during this period (Wang et al. 2018). The mitogenomic results suggest ancient connections between freshwater basins of East Asia and Europe near the Cretaceous–Paleogene boundary, probably via a continuous paleo-river system or along the Tethys coastal line (Hou and Li 2018), and this is also depicted in the Margaritiferinae subfamily within Margaritiferidae (Lopes-Lima et al. 2018b). This pattern is well supported by at least three independent but almost synchronous divergence events: Potomida vs. Lamprotula and Pronodularia, Microcondylaea vs. Solenaia, and Unio vs. Nodularia and its relatives. During the same period, faunal exchange via the Beringian Land Bridge with subsequent vicariance events may also have started. The question of the origin of the family-clade, i.e., Unionidae, remains unanswered due to the lack of available mitogenomes of Parreysiinae and Rectidentinae, although combined COI, 28 S and 16 S data indicated that this family most likely originated within East or Southeast Asia (Bolotov et al. 2017a).
The new results presented herein support the hypothesis that several of the largest river basins on Earth may represent so-called ancient (long-lived) rivers, the Unionida faunas of which have existed throughout long-term periods comparable with geological epochs (Bolotov et al. 2017a; Lopes-Lima et al. 2018b). The mitogenomic results suggest that the MRCA of the entire Gonideinae+Unioninae clade may have originated within the paleo-Yangtze drainage basin. This indicates that the modern Yangtze may be a system of at least Late Jurassic origin and a stable refugium for very ancient, relic lineages that have existed for approximately 150 Ma. The unionid fauna of the paleo-Mississippi system seems to be of Early Cretaceous origin (mean age of the crown group in our model) that has diversified for at least 120 Ma. The paleo-Mekong fauna appears to be younger as it likely separated from the paleo-Yangtze fauna in the Albian - Cenomanian, and its two largest monophyletic unionid radiations may have had a Late Cretaceous or Paleocene origin (Bolotov et al. 2017a, 2017b). These results agree with the dating of divergence between two primary clades of the Southeast Asian cave spitting spiders that were separated at ∼55 Ma by the paleo-Mekong River, which served as a biogeographic barrier (Luo and Li 2017).
Systematics
Based on the morphological evidence, we propose the putative MRCA of the Unionidae and Margaritiferidae as a new fossil family-level taxon in the Unionoidea.
Superfamily Unionoidea Rafinesque, 1820
Family †Shifangellidae Bolotov, Bogan, Lopes-Lima & Froufe fam. nov.
Type genus: †Shifangella Liu & Luo in Liu (1981)
Diagnosis: The Margaritiferidae and Unionidae are the most conchologically similar families to the †Shifangellidae. However, †Shifangellidae can be distinguished from the Margaritiferidae by having a weakly developed, narrow hinge plate (vs. typically well-developed and rather thick) and a shallow, smooth anterior adductor scar (vs. deep with arborescent-like striations), and from the Unionidae by an elongated Margaritifera-like shell with strongly concave ventral margin (vs. typically straight, rounded or slightly concave).
Distribution: Late Triassic, southwestern China (Sichuan).
Biology: This ancestral family likely had parasitic glochidial larvae similar to its descendants (ancestral state reconstruction, probability 100%).
Comments: Synonymy of the genus †Palaeomargaritifera Ma 1996 (Middle Jurassic, China) with †Shifangella suggested by Fang et al. (2009) most likely erroneous because †Palaeomargaritifera has a well-developed, thick hinge plate, strong pseudocardinal teeth and deep anterior adductor scar with arborescent-like striations supporting its original placement within the Margaritiferidae. The genus †Dianoconcha Guo, 1988 (Middle Jurassic, China), another synonym of †Shifangella proposed by Fang et al. (2009), differs by a subtrapezoid, elongate-elliptical or rhomboid shell. This feature together with a narrow hinge plate and an observable but shallow anterior adductor scar suggest that it most likely belongs to the Unionidae. With respect to their age and diagnostic features mentioned above, †Palaeomargaritifera and †Dianoconcha appear to be the MRCAs of the crown groups of the Margaritiferidae and Unionidae, respectively. The family-level placement of several unionoid genera described from the Early Jurassic of China (e.g., †Pseudomargaritifera Ma 1996 and †Solenoides Ma 1996) is unclear and is in need of further revision; some of them might actually be members of the †Shifangellidae.
Conclusions
All the phylogenies inferred in this study using, for the first time, both the M and F mitogenomes individually and combined support the monophyly of the so-called “problematic” Gonideinae taxa. Moreover, the new mitogenomic results place the Pseudodontinae, as previously described by Bolotov et al. (2017a), as a subclade within the monophyletic Gonideinae in both M- and F-type phylogenies. Additionally, the present work supports the hypothesis of an ancient Mesozoic origin and diversification of the Unionoidea and reveals that each TDRL event was followed by the stable, long-term persistence of a mitochondrial gene order through evolving lineages and corresponds to the first reliable mitogenomic evidence supporting the evolutionary stasis in molecular traits of freshwater mussels. Finally, we propose a new systematics framework with three infrafamilial levels (i.e., subfamilies, tribes, and subtribes) that better explains the evolutionary patterns within the Unionidae. Future application of the phylogenetic mitogenome-based approach outlined here to Parreysiinae, Rectidentinae and Modellnaiinae will be an important step to further resolve current taxonomic classification uncertainties within the Unionidae. Moreover, this study demonstrates the considerable potential for using comparative genomic techniques for unravelling patterns in the tempo, timing and mode of evolutionary processes.
Data archiving
Sequence data have been submitted to GenBank accession numbers: MK994770–MK994777.
References
Araujo R, Schneider S, Roe KJ, Erpenbeck D, Machordom A (2017) The origin and phylogeny of Margaritiferidae (Bivalvia, Unionoida): a synthesis of molecular and fossil data. Zool Scr 46:289–307
Banarescu P (1991) Distribution and dispersal of freshwater animals in North America and Eurasia 2. In: Zoogeography of fresh waters, Aula-Verlag, Wiesbaden, Germany, p 519–1091
Basso A, Babbucci M, Pauletto M, Riginella E, Patarnello T, Negrisolo E (2017) The highly rearranged mitochondrial genomes of the crabs Maja crispata and Maja squinado (Majidae) and gene order evolution in Brachyura. Sci Rep 7(1):4096
Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G et al. (2013) MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol 69:313–319
Bernt M, Merkle D, Middendorf M (2008) An Algorithm for inferring mitogenome rearrangements in a phylogenetic tree. In: Setubal JC, Stoye J, Stadler PF (eds) Comparative Genomics. Springer, Berlin, Heidelberg, p 143–157
Bjerrum CJ, Surlyk F, Callomon JH, Slingerland RL (2001) Numerical paleoceanographic study of the Early Jurassic Transcontinental Laurasian Seaway. Paleoceanography 16:390–404
Bogan AE, Weaver PG (2012) A new genus and new species of freshwater mussel from the mid Late Triassic rift lakes of eastern North Carolina (Bivalvia: Unionida: cf. Unionidae). Nautilus 126:105–112
Bolotov IN, Vikhrev IV, Bespalaya YV, Gofarov MY, Kondakov AV, Konopleva ES et al. (2016) Multi-locus fossil-calibrated phylogeny, biogeography and a subgeneric revision of the Margaritiferidae (Mollusca: Bivalvia: Unionoida). Mol Phylogenet Evol 103:104–121
Bolotov IN, Vikhrev IV, Kondakov AV, Konopleva ES, Gofarov MY, Aksenova OV et al. (2017a) New taxa of freshwater mussels (Unionidae) from a species-rich but overlooked evolutionary hotspot in Southeast Asia. Sci Rep 7:11573
Bolotov IN, Kondakov AV, Vikhrev IV, Aksenova OV, Bespalaya YV, Gofarov MY et al. (2017b) Ancient River Inference Explains Exceptional Oriental Freshwater Mussel Radiations. Sci Rep 7:2135
Boore JL (2000) The Duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. Springer, Dordrecht p 133–147
Brandt RAM (1974) The non-marine aquatic Mollusca of Thailand. Arch Moll 105:1–423
Breton S, Beaupre HD, Stewart DT, Piontkivska H, Karmakar M, Bogan AE et al. (2009) Comparative mitochondrial genomics of freshwater mussels (bivalvia: unionoida) with doubly uniparental inheritance of mtdna: gender-specific open reading frames and putative origins of replication. Genetics 183:1575–1589
Breton S, Stewart DT, Shepardson SP, Trdan RJ, Bogan AE, Chapman EG et al. (2011) Novel protein genes in animal mtDNA: A new sex determination system in freshwater mussels (Bivalvia: Unionoida)? Mol Biol Evol 28:1645–1659
Combosch DJ, Collins TM, Glover EA, Graf DL, Harper EM, Healy JM et al. (2017) A family-level tree of life for bivalves based on a Sanger-sequencing approach. Mol Phylogenet Evol 107:191–208
Conant GC, Wolfe KH (2008) GenomeVx: simple web-based creation of editable circular chromosome maps. Bioinformatics 24:861–862
Dowton A, Austin AD (1999) Evolutionary dynamics of a mitochondrial rearrangement “hot spot” in the hymenoptera. Mol Biol Evol 16:298–309
Drummond AJ, Ho SYW, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. PLoS Biol 4:e88
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973
Fang ZJ, Chen JH, Chen CZ, Sha JG, Lan X, Wen SX (2009) Supraspecific taxa of the Bivalvia first named, described, and published in China (1927–2007) Univ Kansas Paleontol Contrib New Ser 17:1–157
Flook P, Rowel H (1995) Homoplastic rearrangements of insect mitochondrial tRNA genes. Naturwissenschaften 82:336–337
Fonseca MM, Lopes-Lima M, Eackles MS, King TL, Froufe E (2016) The female and male mitochondrial genomes of Unio delphinus and the phylogeny of freshwater mussels (Bivalvia: Unionida). Mitochondrial DNA Part B 1:954–957
Froufe E, Gan HM, Lee YP, Carneiro J, Varandas S, Teixeira A et al. (2016) The male and female complete mitochondrial genome sequences of the Endangered freshwater mussel Potomida littoralis (Cuvier, 1798) (Bivalvia: Unionidae). Mitochondrial DNA Part A 27:3571–3572
Gan H, Schultz MB, Austin CM (2014) Integrated shotgun sequencing and bioinformatics pipeline allows ultra-fast mitogenome recovery and confirms substantial gene rearrangements in Australian freshwater crayfishes. BMC Evol Biol 14:19
Graf DL (2002) Molecular Phylogenetic Analysis of Two Problematic Freshwater Mussel Genera (Unio and Gonidea) and a Re-Evaluation of the Classification of Nearctic Unionidae (Bivalvia: Palaeoheterodonta: Unionoida). J Moll Stud 68:65–71
Graf DL, Cummings KS (2006) Palaeoheterodont diversity (Mollusca: Trigonioida+Unionoida): What we know and what we wish we knew about freshwater mussel evolution. Zool J Linn Soc 148:343–394
Heard WH, Hanning GW (1978) Anatomical Features of Modellnaia siamensis (Mollusca, Bivalvia, Unionidae). Zool Scr 7:219–223
Hesselbo SP, Robinson SA, Surlyk F, Piasecki S (2002) Terrestrial and marine extinction at the Triassic-Jurassic boundary synchronized with major carbon-cycle perturbation: A link to initiation of massive volcanism? Geology 30:251–254
Hou Z, Li S (2018) Tethyan changes shaped aquatic diversification. Biol Rev 93:874–896
Huang X-C, Rong J, Liu Y, Zhang M-H, Wan Y, Ouyang S et al. (2013) The complete maternally and paternally inherited mitochondrial genomes of the endangered freshwater mussel solenaia carinatus (bivalvia: unionidae) and implications for unionidae Taxonomy. PLoS ONE 8:e84352
Huang X-C, Ouyang S, Ouyang J, Zhou S, Wu XP (2019) Towards a global phylogeny of freshwater mussels (Bivalvia: Unionida): Species delimitation of Chinese taxa, mitochondrial phylogenomics, and diversification patterns. Mol Phylogenetics Evol 130:45–59
IUCN (2019). The IUCN Red list of threatened species. version 2019-1. http://www.iucnredlist.org. Accessed Jan 2019
Jacobsen MW, Pujolar JM, Gilbert MTP, Moreno-Mayar JV, Bernatchez L, Als TD et al. (2014) Speciation and demographic history of Atlantic eels (Anguilla anguilla and A. rostrata) revealed by mitogenome sequencing. Heredity 113:432–442
Katoh K, Standley DM (2013) MAFFT Multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Konopleva ES, Bolotov IN, Vikhrev IV, Gofarov MY, Kondakov AV (2017) An integrative approach underscores the taxonomic status of Lamellidens exolescens, a freshwater mussel from the Oriental tropics (Bivalvia: Unionidae). Syst Biodiv 15:204–217
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B (2016) PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol 34:772–773
Laslett D, Canbäck B (2008) ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24:172–5
Liu X-Z (1981) On some newly discovered non-marine pelecypods from the Late Triassic Wuzhongshan Formation in Sichuan Basin. Bull Chengdu Inst Geol Min Resour Chin Acad Geol Sci 2:121–136
Liu Q-N, Xin Z-Z, Bian D-D, Chai X-Y, Zhou C-L, Tang B-P (2016) The first complete mitochondrial genome for the subfamily Limacodidae and implications for the higher phylogeny of Lepidoptera. Sci Rep 6:35878
Lopes-Lima M, Teixeira A, Froufe E, Lopes A, Varandas S, Sousa R (2014) Biology and conservation of freshwater bivalves: past, present and future perspectives. Hydrobiologia 735:1–13
Lopes-Lima M, Froufe E, Do VT, Ghamizi M, Mock KE, Kebapçı Üet al (2017a) Phylogeny of the most species-rich freshwater bivalve family (Bivalvia: Unionida: Unionidae): Defining modern subfamilies and tribes Mol Phylogenet Evol 106:174–191
Lopes-Lima M, Fonseca MM, Aldridge DC, Bogan AE, Gan HM, Ghamizi Met al (2017b) The first Margaritiferidae male (M-Type) mitogenome: Mitochondrial gene order as a potential character for determining higher-order phylogeny within Unionida (Bivalvia) J Moll Stud 83:249–252
Lopes-Lima M, Burlakova LE, Karatayev AY, Mehler K, Seddon M, Sousa R (2018a) Conservation of freshwater bivalves at the global scale: diversity, threats and research needs. Hydrobiologia 810:1–14
Lopes-Lima M, Bolotov IN, Do VT, Aldridge DC, Fonseca MM, Gan HM et al. (2018b) Expansion and systematics redefinition of the most threatened freshwater mussel family, the Margaritiferidae. Mol Phylogenet Evol 127:98–118
Luo Y, Li S (2017) Cave Stedocys spitting spiders illuminate the history of the Himalayas and southeast Asia. Ecography 41:414–423
Ma Q (1996) Revision of Mesozoic Margaritiferidae in China and their development. Acta Palaeontol Sin 35:408–429
Miller MA, Pfeiffer W, Schwartz T (2010). Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: 2010 Gateway Computing Environments Workshop (GCE), IEEE. p 1–8
Moritz C, Dowling TE, Brown WM (1987) Evolution of animal mitochondrial DNA: relevance for population biology and systematics. Annu Rev Ecol Syst 18:269–292
Percival LME, Ruhl M, Hesselbo SP, Jenkyns HC, Mather TA, Whiteside JH (2017) Mercury evidence for pulsed volcanism during the end-Triassic mass extinction. Proc Natl Acad Sci USA 114:7929–7934
Pereira SL (2000) Mitochondrial genome organization and vertebrate phylogenetics. Genet Mol Biol 23:745–752
Pfeiffer III JM, Graf DL (2013) Re-analysis confirms the polyphyly of Lamprotula Simpson, 1900 (Bivalvia: Unionidae). J Moll Stud 79:249–256
Pfeiffer III JM, Graf DL (2015) Evolution of bilaterally asymmetrical larvae in freshwater mussels (Bivalvia: Unionoida: Unionidae). Zool J Linn Soc 175:307–318
Pfeiffer JM, Breinholt JW, Page LM (2019) Unioverse: a phylogenomic resource for reconstructing the evolution of freshwater mussels (Bivalvia, Unionoida). Mol Phylogenet Evol 137:114–126
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA, Susko E (2018) Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst Biol 67:901–904
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S et al. (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Sela I, Ashkenazy H, Katoh K, Pupko T (2015) GUIDANCE2: accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res 43:W7–W14
Schneider S, Böhme M, Prieto J (2013) Unionidae (Bivalvia; Palaeoheterodonta) from the Palaeogene of northern Vietnam: exploring the origins of the modern East Asian freshwater bivalve fauna. J Syst Palaeontol 11:337–357
Scholz H, Glaubrecht M (2004) Evaluating limnic diversity: Toward a revision of the unionid bivalve Coelatura Conrad, 1853 in the Great Lakes of East Africa and adjacent drainage systems (Mollusca, Bivalvia, Unionidae). Zoosyst Evol 80:89–121
Schwarz G (1978) Estimating the dimension of a model. Ann Stat 6:461–464
Smithwick FM, Stubbs TL (2018) Phanerozoic survivors: Actinopterygian evolution through the Permo-Triassic and Triassic-Jurassic mass extinction events. Evolution 72:348–362
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Starobogatov YI (1970) Fauna Mollyuskov i Zoogeograficheskoe Raionirovanie Kontinental’nykh Vodoemov Zemnogo Shara [Mollusk Fauna and Zoogeographical Partitioning of Continental Water Reservoirs of theWorld]. Nauka, Leningrad, Russia
Taylor DW (1988) Aspects of freshwater mollusc ecological biogeography. Palaeogeogr Palaeoclim Palaeoecol 62:511–576
Van Damme D, Bogan AE, Dierick M (2015) A revision of the Mesozoic naiads (Unionoida) of Africa and the biogeographic implications. Earth-Sci Rev 147:141–200
Wang P, Zheng H, Liu S, Hoke G (2018) Late Cretaceous drainage reorganization of the Middle Yangtze River. Lithosphere 10:392–405
Watters TG (2001) The Evolution of the Unionacea in North America, and its implications for the worldwide fauna. In: Bauer G, Wachtler K (eds) Ecology and evolution of the freshwater Mussels Unionoida. Ecological Studies, vol. 145. Springer-Verlag, Berlin, pp 281–307
Wesselingh FP (2007) Long-Lived Lake Molluscs as Island Faunas: A Bivalve Perspective. Springer, Dordrecht, pp 275–314
Whelan NV, Geneva AJ, Graf DL (2011) Molecular phylogenetic analysis of tropical freshwater mussels (Mollusca: Bivalvia: Unionoida) resolves the position of Coelatura and supports a monophyletic Unionidae. Mol Phylogenet Evol 61:504–514
Wu RW, Liu XJ, Wang S, Roe KJ, Ouyang S, Wu XP (2019) Analysis of mitochondrial genomes resolves the phylogenetic position of Chinese freshwater mussels (Bivalvia, Unionidae). ZooKeys 812:23
Yu Y, Harris AJ, Blair C, He X (2015) RASP (Reconstruct Ancestral State in Phylogenies): a tool for historical biogeography. Mol Phylogenet Evol 87:46–49
Yuan Y, Li Q, Yu H, Kong L (2012) The complete mitochondrial genomes of six heterodont bivalves (Tellinoidea and Solenoidea): variable gene arrangements and phylogenetic implications. PLoS ONE 7:e32353
Zouros E, Oberhauser Ball A, Saavedra C, Freeman KR (1994) An unusual type of mitochondrial DNA inheritance in the blue mussel Mytilus. Proc Natl Acad Sci USA 91:7463–7467
Acknowledgements
The authors wish to thank the Editor and the three anonymous reviewers for helpful remarks and suggestions that improved the quality of the manuscript. This research was developed under ConBiomics: the missing approach for the Conservation of freshwater Bivalves Project N° NORTE-01-0145-FEDER-030286, co-financed by COMPETE 2020, Portugal 2020 and the European Union through the ERDF, and by FCT through national funds (UID/Multi/04423/2019). FCT also supported MLL (SFRH/BD/115728/2016). The Russian Ministry of Education and Science (project no. 6.2343.2017/4.6), the Federal Agency for Scientific Organizations (project no. 0409-2015-0143), the Presidium of the Russian Academy of Sciences (scientific program no. 52), and the Russian Foundation for Basic Research, RFBR (project no. 17-45-290066) supported INB. Permits for fieldwork and sampling in Malaysia were issued by the Malaysian Ministry of Higher Education (FRGS/1/2015/WAB13/UNIM//1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Froufe, E., Bolotov, I., Aldridge, D.C. et al. Mesozoic mitogenome rearrangements and freshwater mussel (Bivalvia: Unionoidea) macroevolution. Heredity 124, 182–196 (2020). https://doi.org/10.1038/s41437-019-0242-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-019-0242-y
This article is cited by
-
No evidence of DUI in the Mediterranean alien species Brachidontes pharaonis (P. Fisher, 1870) despite mitochondrial heteroplasmy
Scientific Reports (2022)
-
Phylogeny and biogeography of Indochinese freshwater mussels in the genus Pilsbryoconcha Simpson, 1900 (Bivalvia: Unionidae) with descriptions of four new species
Scientific Reports (2022)
-
Mitogenomic architecture of the multivalent endemic black clam (Villorita cyprinoides) and its phylogenetic implications
Scientific Reports (2020)
-
New freshwater mussel taxa discoveries clarify biogeographic division of Southeast Asia
Scientific Reports (2020)
