
Abstract
Mutations conferring loss of function at the FLNA (encoding filamin A) locus lead to X-linked periventricular nodular heterotopia (XL-PH), with seizures constituting the most common clinical manifestation of this disorder in female heterozygotes. Vascular dilatation (mainly the aorta), joint hypermobility and variable skin findings are also associated anomalies, with some reports suggesting that this might represents a separate syndrome allelic to XL-PH, termed as Ehlers-Danlos syndrome-periventricular heterotopia variant (EDS-PH). Here, we report a cohort of 11 males and females with both hypomorphic and null mutations in FLNA that manifest a wide spectrum of connective tissue and vascular anomalies. The spectrum of cutaneous defects was broader than previously described and is inconsistent with a specific type of EDS. We also extend the range of vascular anomalies associated with XL-PH to included peripheral arterial dilatation and atresia. Based on these observations, we suggest that there is little molecular or clinical justification for considering EDS-PH as a separate entity from XL-PH, but instead propose that there is a spectrum of vascular and connective tissues anomalies associated with this condition for which all individuals with loss-of-function mutations in FLNA should be evaluated. In addition, since some patients with XL-PH can present primarily with a joint hypermobility syndrome, we propose that screening for cardiovascular manifestations should be offered to those patients when there are associated seizures or an X-linked pattern of inheritance.
Similar content being viewed by others


Introduction
Periventricular heterotopia (PH) results from abnormal radial neuronal migration during late embryonic and early fetal development in which morphologically normal neurons are located in an anatomically abnormal position lining the lateral margins of the ventricles. PH is usually diagnosed when patients present with a seizure disorder and neuroimaging studies demonstrate heterotopic gray matter in a periventricular distribution. PH is a genetically and phenotypically heterogeneous disorder.1, 2 The most commonly identified genetic cause, representing fewer than half of all PH cases, is a mutation in the X-linked gene FLNA (XL-PH; OMIM #300049). The substantial majority of individuals with FLNA-related PH are females since hemizygosity for null mutations characteristically leads to male embryonic lethality. Mutations that give rise to these conditions are qualitatively different from those that lead to a spectrum of skeletal dysplasias termed as the otopalatodigital syndrome spectrum disorders.3
Two previous reports linked XL-PH to a connective tissue disorder characterized by joint hypermobility, cutaneous anomalies and aortic aneurysms (Ehlers-Danlos syndrome (EDS)-PH; OMIM #300537), and reported three sporadic and one familial case of the association.4, 5 The clinical manifestations of this syndrome are poorly defined since the detailed descriptions of some reported cases are not available. It is also unclear if the four FLNA mutations that underlie EDS-PH are functionally different from those that cause generic XL-PH. In addition, it is uncertain whether cardiac valvular defects, proximal aortic aneurysmal disease, joint hypermobility and cutaneous anomalies coexist with sufficient regularity to be considered as a separate diagnostic entity. Here, we describe 11 patients presenting with PH and a spectrum of connective tissue abnormalities characterized by combinations of vascular, cardiac, cutaneous and joint-related symptoms and signs due to mutations in FLNA. The responsible mutations in FLNA constitute null alleles in female patients, while males present with a broader spectrum of connective tissue abnormalities and harbor hypomorphic FLNA mutations. These observations highlight that variability in the extra-cerebral manifestations of XL-PH is not limited to females. We propose that loss of function mutations in FLNA confer a spectrum of pleiotropic phenotypic effects that are both broader and qualitatively different than those typically seen in EDS, suggesting that the association of PH with these anomalies may not warrant recognition as a separate syndrome.
Materials and methods
In the present study, we evaluated two families and seven sporadic individuals with XL-PH following physician-initiated referral. Informed consent was obtained under approvals granted by the New Zealand Multiregional Ethics Committee and the Cedars-Sinai IRB (protocols 0463 and 4232). These individuals have not been reported previously. Sequencing of FLNA and measurement of the degree of skewing of X-inactivation utilizing methylation-sensitive PCR were performed as previously described.6 Mutations are denoted according to the reference sequence NM_001456.1. Immunohistochemical staining of skin fibroblasts and western analysis of lymphoblastoid cell lines utilized anti-FLNA (MAB1678, Chemicon International, Billerica, MA, USA) and anti-FLNB (AB9276, Chemicon International) antibodies.
Results
The diagnosis of XL-PH in all patients was established by neuroimaging undertaken for a range of indications including seizures, developmental delay, muscle weakness, family history of PH and other neurological manifestations. In all patients, subsequent studies demonstrated a pathogenic mutation in FLNA. The pertinent connective tissue abnormalities, imaging findings and mutations are summarized in Table 1.
Clinical reports
Cases F1 and F2
The proband (F1) is a 38-year-old female previously diagnosed with a sinus of Valsalva aneurysm, necessitating a valve-sparing aortic root repair (Figure 1). The histopathology of the excised aortic tissue revealed areas of mild cystic medial degeneration. A previous MRI of the brain, performed because of steroid-induced myopathy, revealed bilateral PH and head, thoracic and abdominal MRI/A studies demonstrated diffuse ectasia of aortic branch vessels. She has always been loose jointed and reports a history of clumsiness during childhood but no joint dislocations or fractures. She reports easy bruising, her skin is soft but not hyperextensible, and she has a normal scar formation. Electron microscopy studies of her skin revealed a normal pattern of collagen and elastic fibers. Her 19-year-old daughter (F2) is loose jointed, bruises easily and has soft skin albeit with normal elasticity (Figure 1). Further imaging studies in F2 revealed a normal aorta but an aneurysm of the proximal right subclavian artery up to 3.2 cm, and bilateral widespread neuronal heterotopia, similar to her mother. Sequencing of FLNA in F1 revealed an 8-bp deletion in exon 6, c.883_890 del8, which predicts a frameshift and premature truncation of translation (p.Asn296Glu fs*38). Targeted sequencing analysis confirmed the presence of the same mutation in F2, but not in her sister that did not exhibit any connective tissue abnormalities. X-inactivation studies on DNA from blood and cultured skin fibroblasts showed ratios indicative of random inactivation. Immunohistochemical staining of cultured fibroblast using anti-FLNA antibody demonstrated two cell populations, one expressing FLNA in a normal distribution and another that had no identifiable FLNA protein. Sequencing of cDNA obtained from these cells demonstrated sequence only from the non-mutation bearing allele and RT-PCR study using primers located over sequences encoding exon boundaries 4/5 and 9/10 showed no evidence for exon skipping induced by the mutation. It was therefore concluded that this mutation constitutes a null FLNA allele.

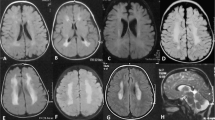
Cases F1 and F2. (a, c) Periventricular heterotopia in F1 and F2; (b) small joint hypermobility in F1; (d) Small and large joint hypermobility in F2; (e) 3D chest MRI reconstruction demonstrating an aneurysm of the proximal right subclavian artery in F2; (f) Immunostaining for filamin A, filamin B and merged images in cells from F2 (upper row) and control cells (lower row). There is a cell population in F2 that does not stain for filamin A but stains for filamin B and represents cells expressing the mutation-bearing allele.
Case F3
This 6-year-old female underwent a neonatal echocardiogram because of a cardiac murmur that showed extreme dilatation of the main pulmonary artery and its branches, ascending and descending aorta and head and neck branches of the aorta (Figure 2). In addition, the pulmonary, tricuspid and aortic valves were dysplastic. A large persistent ductus arteriosus was surgically ligated at 10 weeks of age. At 6 years, her valvular dysplasia persisted, but the ectasia of the ascending aorta and main pulmonary artery demonstrated no progression. Pulmonary artery pressure was at systemic levels, even after ligation of the ductus arteriosus. She had mild cyanosis due to a right to left shunt at the atrial level. This was reversible with oxygen supplementation, but her pulmonary arterial pressures remain abnormally reactive, and when she is upset, they increase significantly. She was treated with sildenifil and oxygen supplementation until age 18 months and is still on sildenifil. Attempts to discontinue treatment are associated with recurrence of pulmonary hypertension, the cause of which remains unknown, although there is coexistent lung disease with areas of focal hyperinflation associated with minimal patchy atelectasis evident on her chest radiographs. A brain MRI scan performed at age 3 weeks demonstrated bilateral PH. Neurological examination and psychomotor development were initially normal, with no seizures, but at age 6 she has delayed development of speech and language compared with her peers. She has social shyness and anxiety but cognitive development, although not tested formally, appears to be within the normal range. Although skin and joints were initially normal, generalized joint laxity was evident by 2½ years and this has persisted. Her skin is translucent with increased venous patterning, but not hyperelastic. She was shown to be heterozygous for a de novo truncating FLNA mutation c.2193C>A that predicts (p.Tyr731X).

Cases F3 (a–d), F4 (e) and F5 (f–h), and M1 (i). (a) Axial MRI scan demonstrating PH (arrows). (b) Transthoracic echocardiography demonstrating dilatation of the aorta. (c) Redundant skin folds at the umbilicus. (d) Caudal appendage. (e) MRI scan demonstrating PH. (f) Dilated ascending, transverse and descending aorta. (g) Thin translucent skin. (h) Small joint hypermobility. (i) Western analysis of lymphoblastoid cell lysates obtained from case M1 for FLNA protein expression. FLNA abundance was 30% (SD±8) compared with control cell lines after normalization to GAPDH expression.
Case F4
This patient is an 18-year-old female, born at term after a normal dichorionic, diamniotic twin pregnancy (Figure 2). A neonatal echocardiogram revealed a VSD, which was surgically repaired although she remains under bosentan treatment for persistent pulmonary hypertension. At 2 years, she presented with generalized seizures. A brain MRI demonstrated bilateral symmetrical PH. She walked at 2 years and speech was delayed. At age 12 years, an echocardiogram demonstrated pulmonary arterial root dilatation. Generalized joint laxity was evident with a Beighton score of 7/9. Her skin was thin with visible vessels. Sequencing of FLNA revealed no mutations but MLPA analysis demonstrated a de novo FLNA deletion including exons 2, 5 and 13. Probes located in the EMD gene, which is located 5' to FLNA, and in exon 15 showed a normal copy number.
Case F5
The proband is a 23-year-old woman. After a normal term pregnancy and delivery, an umbilical hernia was repaired at age 2 (Figure 2). She also had transient bilateral congenital hip dysplasia, treated conservatively. There was no history of motor delay, but she continued to subluxate her hip throughout childhood. Peripheral cyanosis at the age of 7 years led to a cardiac assessment, and aortic valve regurgitation was identified followed by progressive aortic root dilatation up to 4.5 cm at the sinuses of Valsalva, with a dilated proximal aorta and pulmonary artery. At age 16 she had her first seizure, and brain MRI showed PH. Clinical assessment at age 23 showed joint hypermobility (Beighton score 8/9), mild skin thinning at the proximal upper limbs, ‘criss-crossing’ of the palmar skin and broad feet. FLNA analysis revealed that she is heterozygous for a single nucleotide deletion c.7813delC (p.Leu2605fs) leading to a termination of translation within the dimerization domain of the FLNA protein. Her mother, who demonstrated joint hypermobility, did not carry the FLNA mutation. Light and electron microscopy of skin punch biopsies showed normal collagen packing
Case F6
The patient presented for genetics evaluation at 15 years due to a history of complex partial seizures and mild developmental delays. She was born at 32 weeks gestation via spontaneous vaginal delivery following premature onset of labor. A prenatal ultrasound showed ventriculomegaly with no other abnormalities. The presence of macrocephaly prompted brain imaging at 8 months of age that demonstrated ventriculomegaly, agenesis of the corpus callosum and bilateral periventricular gray matter heterotopia. She had a febrile seizure at 11 months of age and a second a febrile seizure at 17 months. Complex partial seizures persisted throughout childhood with trials of several anti-seizure medications. She underwent implantation of a vagus nerve stimulator at 13 years. She has mild mitral valve regurgitation with no structural abnormalities of the mitral valve.
In elementary school she received academic support for math and reading, she is now in high school in typical classroom with extra-tutoring for English. Both parents have mitral valve prolapsed and the mother self-reported a history of hyper-mobile joints but no seizures. Physical exam at the time of initial evaluation showed height of 155.2 cm (15th percentile), weight of 41.0 kg (5th percentile), head circumference of 57.25 cm (90th percentile), hyper-mobile joints in the upper and lower limbs, ocular motor apraxia and ocular tics. Sequence analysis of FLNA revealed heterozygosity for a maternally inherited c.1829-2 A>G mutation, predicted to interfere with the consensus acceptor slice site of intron 12.
Cases F7 and M1
The proband is a 10-year-old male. Medical history includes a small atrial septal defect that was noted at 1 month and a supra-umbilical hernia was repaired at 8 years. At age 10, a follow-up echocardiogram demonstrated a prolapse of the mitral valve. Shortly afterwards, he presented with partial seizures and brain MRI scan showed PH with no other brain structural anomalies. He has a normal body habitus, with thin and translucent skin, which is not hyperelastic and scar healing is normal. His 41-year-old mother presented with partial and generalized seizures at the age of 24 years and a brain MRI showed PH. The pregnancy was notable for maternal carbamazepine treatment, which was maintained throughout gestation. The mother has thin skin that is not hyperelastic and an echocardiogram revealed normal heart. Sequencing of FLNA showed that the mother is heterozygous, and the son is hemizygous, for the mutation c.853C>T that predicts an amino-acid substitution p.Arg285Cys. Western blot analysis of transformed lymphoblastoid cells from the son demonstrated a ratio of expression of FLNA protein compared with a control cell line obtained from a gender and age matched control of 0.30±0.08 (±SEM).
Case M2
This 2-month-old male was born at term, following an uneventful pregnancy with normal prenatal ultrasound scans (Figure 3). He had normal weight, length and head circumference for gestational age. Two hours after delivery he became cyanotic and tachypneic, and an echocardiogram showed marked dilatation of the ascending aorta with a dysplastic aortic valve, atrial septal defect, small ventricular septal defect and patent ductus arteriosus. Brain MRI showed hypoplasia of the corpus callosum, PH and cervical syringomyelia. Clinical examination showed normal body proportions, marked joint laxity and increased skin elasticity. Further imaging studies showed marked dilatation of the whole length of the thoracic and abdominal aorta, elongated and tortuous supra-aortic vessels, and enlarged pulmonary arteries. At the bifurcation of the iliac arteries, there was a persistence of the embryonic femoral artery with anomalous persistence of the embryonic common right external iliac artery. He developed intractable pulmonary hypertension with heart failure and eventually died at 2 months due to sepsis. Additional findings at post mortem included thickened mitral and tricuspid valves and intestinal malrotation. Electron microscopy examination of the brachiocephalic truncus media showed fragmentation and loss of the elastic lamellae creating ‘cyst-like’ spaces, and fiber disarray with loss of parallel organization of the elastic fibers. These changes, although milder, were also observed in the dilated portions of the inner media of the aortic wall. Sequencing of FLNA revealed hemizygosity for a de novo c.5498_5504delCACCCACinsAC indel mutation. The translational consequence of this mutation is unclear and no cells were available for transcript analysis.

Case M2. (a–d) 3D reconstruction from MDCT angiogram. (a) Right posterolateral view of the thoracic aorta. Note the enlarged and elongated supra-aortic arteries and the pulmonary arteries (*) with a relatively conserved diameter. (b) Posterior view of the descending aorta and the persistent ductus arteriosus (PDA). Common origin of the right brachiocephalic trunk (RBT) and the left carotid artery (LCA). (c, d) Axial and oblique MIP reconstructions demonstrating an enlarged PDA and pulmonary arteries. (e, f) VRT 3D reconstruction demonstrating right (e) and left (f) anterior coronal views of the abdominal aorta and pelvic vessels. The celiac trunk (CT), superior mesenteric artery (SMA) and inferior mesenteric artery (IMA) are normal. The abdominal aorta terminates as a tapering sac (*) from which many small caliber vessels emanate (arrows). (g, h) Coronal and sagittal MIP reconstructions of the pelvic vessels and abdominal aorta showing a progressive decrease of the diameter distally. (i) Coronal section of the brain demonstrating heterotopic nodules (arrowed) in a periventricular distribution. (j) Nodular and thickened mitral and tricuspid valves. (k, l) Microscopic cross-sections of the brachiocephalic artery media showing fragmentation and/or loss of the elastic lamellae creating ‘cyst-like’ spaces, thinner lamellae with greater distances between them and loss of parallel organization of the elastic fibers (k: H&E; l: Van Gieson’s stain).
Case M3
This 15-year-old male patient was diagnosed with EDS at age 5 on the basis of soft and slightly hyperextensible skin, diaphragmatic eventration, an umbilical hernia and joint hypermobility (Beighton score 8/9). He also had mild pectus carinatum, and mild lumbar scoliosis. His palate and uvula were normal. He had normal wound healing, normal scarring, normal bruising and no joint dislocations. Electron microscopy and elastin staining of a skin biopsy sample were normal. Paucinodular PH was diagnosed on MRI at age 15 during investigation for headaches and episodes of dizziness and poor balance. There were no associated cortical malformations. His early childhood development, subsequent learning, intelligence and mainstream school progress were normal. Echocardiogram showed mild mitral valve prolapse with thickened mitral valve leaflets and a normal aortic valve. The aortic diameter at the level of the sinuses of Valsalva was 31 mm (normal range <28 mm). MRA of the cerebral arteries was normal. At 16 years he was admitted because of a spontaneous pneumothorax. FLNA analysis showed that he was hemizygous for a maternally inherited c.381G>C mutation predicting a p.Lys127Asn substitution. His mother demonstrates mild joint hypermobility with a Beighton score of 4/8 and no neurological symptomatology. The maternal grandparents did not have the c.381G>C mutation, indicating that it had arisen de novo in the mother.
Case M4
A 1-year-old male, presented with cutis-laxa and dysplastic mitral and tricuspid valves in the early neonatal period (Figure 4). He is the second child born to unrelated parents. His mother and sister have no history of joint laxity, seizures or cutaneous anomalies. The pregnancy had been complicated by maternal spinal tuberculosis diagnosed in the third trimester. Intrauterine growth retardation and a dilated cisterna magna were noted on antenatal ultrasound exam at 22 weeks of gestation and cardiomegaly with mitral and tricuspid valve regurgitation was observed at 28 weeks. He was born at 36 weeks gestation weighing 2.57 kg and developed respiratory distress requiring oxygen supplementation for the first few days of life. Postnatal echocardiogram found severely dysplastic mitral and tricuspid valves, which progressed over the first year of life to lead to cardiac failure and recurrent pulmonary infections, necessitating intermittent ventilation and inotropic support. He underwent mitral and tricuspid valvuloplasty at 10 months of age. MRI of the brain in the neonatal period showed PH. He has markedly lax skin, particularly of the palms and soles with increased joint laxity. He has a high palate and a caudal appendage. He underwent repair of a right inguinal hernia at 2 months of life. Wound healing has been normal. At 1 year of age, he remains hypotonic and is fed via nasogastric tube. He has had no seizures. Genetic evaluation including karyotype and array CGH were normal and sequencing of FLNA revealed a maternally inherited c. 387C>G mutation, which predicts p.Ile129Met.

Case M4. (a–c) Cutis laxa and caudal appendage with wrinkling of the skin more evident in the hands and feet.
Discussion
Connective tissue manifestations including joint hypermobility, arterial dilatation, cardiac valvular defects and, to lesser extent, cutaneous anomalies, are well-established associations with XL-PH due to mutations in FLNA.7, 8, 9 However, the grounds for ascribing a separate syndrome assignation (EDS-PH) to the presentation of PH with joint hypermobility (with or without cutaneous anomalies) associated with FLNA mutations are not clear.
EDS-PH syndrome was defined by Sheen et al4 in a report of three unrelated females with mutations (one missense, two frameshift) in FLNA and clinical features of a connective tissue disorder. Nine additional sporadic and two familial occurrences of PH with associated connective tissue abnormalities had no demonstrable mutation in FLNA. Most patients in this cohort were ascertained because of a seizure disorder. Two FLNA mutation-positive females had aortic aneurysms while the third had myxomatous changes in the mitral and aortic valves. All had some degree of joint hypermobility but the clinical description of the connective tissue involvement was brief and therefore difficult to correlate with well-delineated EDS subtypes. It was further proposed that PH may represent a disorder of impaired periventricular intercellular or cell-matrix adhesion and the authors postulated that aortic dilatation and hyperextensible skin observed in EDS-PH may have similar pathological underpinnings.10 Gómez-Garre et al5 reported EDS-PH due to an FLNA mutation (p.Ala128Val), in females in a pedigree presenting with seizures and joint hypermobility, but they reported no cardiovascular involvement except for mitral valve prolapse. Solé et al7 screened patients presenting with PH for mutations in FLNA and identified 15 carriers out of 32 screened. Four additional patients were diagnosed with EDS (one with no identifiable mutation in FLNA), based on their medical records, although patients manifested variable skin findings and joint complications. Cardiovascular manifestations included aortic aneurysms in all three patients and aortic valve insufficiency in one. de Wit et al11 described a cohort of five patients with XL-PH with left sided cardiac valvular or aortic anomalies, but only one male had cutaneous and joint anomalies. A recent study of an Asian family with similar vascular defects and PH reported no accompanying joint hypermobility or skin anomalies; however, no FLNA studies were pursued.12
Our data extend these previous observations in several ways. First, we show that the spectrum of connective tissue abnormalities in XL-PH is much broader than previously reported. Although some patients present with rather benign hypermobility syndrome (cases F6, F7, M3), others manifest life-threatening disorders with variable combinations of vascular, cardiac, pulmonary and joint-related symptoms and signs. Second, we show that dilatation of the aorta is not the only phenotypic component of XL-PH, but arterial disease can be diffuse and manifest in the peripheral vasculature such as in the subclavian artery (F2), in the pulmonary arterial tree (F3) and as an atretic malformation of the distal aorta in a male (M2). Third, we catalog significant variability in the cutaneous manifestations of XL-PH, inconsistent with a specific EDS subtype. Fourth, we demonstrate that two mutations (found in cases F1–4) unequivocally constitute null alleles, and that a missense mutation (F6 and M1) was associated with both PH and joint hypermobility due to reduced expression of FLNA protein (Figure 1). XL-PH in female heterozygotes results principally from null alleles, although missense alleles can result in PH presenting in both males and females.8, 13 Our demonstration of both null and hypomorphic alleles resulting in XL-PH-connective tissue-vascular presentations suggests that there is little at the molecular level to separate the mutations underlying these presentations from those reported to lead to undifferentiated XL-PH in large, previously reported, cohorts.14, 15
The mutations identified in subjects F1–F6 are non-sense, splice site or frameshift mutations that, if they do not lead to non-sense mediated mRNA decay (F1; Figure 1f), are otherwise predicted to produce proteins missing key domains such as integrin binding sites and the homodimerization domain in FLNA. They can therefore be confidently considered to be functionally null alleles. The mutations leading to disease in males are likely to be hypomorphic since some function likely needs to be preserved to permit survival in hemizygotes. The mutation in M1 results in a reduction in quantity of FLNA protein (Figure 2i), while the consequences of the complex indel found in M2 are hard to predict although the fact that it has arisen de novo makes it very likely to be pathogenic. The two missense mutations in M3 and M4 are harder to interpret since in one instance the p.Lys127Asn mutation is non-penetrant in the mother and for p.Ile129Met no data were available on the parents. However, both substitutions not only occur in the CH1 domain of the actin binding site of FLNA but they also lie within a mapped contact point for actin, termed as actin binding site 2. Strengthening the case for pathogenicity is the observed conservation of Ile129 in other actin binding domains in the human (filamin B, filamin C, β-spectrin, dystrophin and α-actinin-4) while Lys127 is likewise conserved in filamin B and filamin C). It must be emphasized that even though 3/11 individuals described in this report are male, this is not indicative of an increased representation of males with XL-PH-connective tissue manifestations above that which would be expected in XL-PH in general. The cohort described here was not ascertained so that it is numerically representative of this presentation amongst the ∼50 unrelated individuals (from a total of 200 individuals screened with PH) that we have diagnosed with FLNA mutations causative of PH at our center.
Stemming from our observations we propose that EDS-PH does not warrant recognition as a separate EDS subtype, but rather, the ‘EDS’ label describes connective tissue abnormalities that are part of the clinical spectrum of XL-PH. We suggest that although individuals may share the same FLNA mutation (F1/F2 and F6/M1) and consequences at the protein level (cases F1–4 all have null alleles) with the same pattern of X-chromosome inactivation (Table 1), they may still exhibit divergent connective tissue abnormalities, cognitive profiles and outcome. This variable expressivity of XL-PH is therefore unlikely to be explained by allelic heterogeneity and consequently constitutes another reason why a separate nosological entity for those individuals demonstrating extremes of phenotypic presentation is not warranted.
This study examines the cutaneous manifestations of XL-PH in more detail than previously reported cases and demonstrates variable findings in the skin in males and females with XL-PH. Most patients demonstrated minimal involvement of the skin with no quantitative or structural changes on skin histology including EM studies (F1, F2, F3, F4, M3, M4), reminiscent of the findings observed in the hypermobility type of EDS.16 The other variable skin presentations were observed in single or familial cases emphasizing the heterogeneity in the cutaneous manifestations (criss cross patterning in F5, thin-translucent skin in M1 and cutis laxa in M2 and M4). Notably, the histopathological findings in M2 (fragmentation and disarray of elastic fibers) added to the recently reported abnormalities in bronchial artery cartilage in patients with XL-PH and lung emphysema,17, 18 may suggest that FLNA deficiency affects elastin fiber integrity. Ferland et al19 demonstrated that FLNA is expressed in the progenitor basal layer of the epidermis, but how this observation relates to the clinical findings described here, including elastin fiber disarray, will require further mechanistic study. The variable skin findings in our cohort certainly warrant a complete evaluation of skin, joints and vasculature in each affected patient with XL-PH.
Vascular involvement has been described mainly in vascular EDS (type IV; Pepin et al20), although arterial fragility occasionally has been reported in classical,21 and kyphoscoliotic EDS.22 Non-progressive aortic root dilatation has occasionally been observed in EDS-hypermobility type.23 The vascular manifestations in XL-PH are different from those observed in the hypermobility and classic types of EDS; they can be more extensive, progressive and may involve peripheral arteries. A recently delineated, rare form of EDS termed as dermatan 4-O-sulfotransferase 1-deficient EDS (D4ST1-deficient EDS), which is characterized by multi-system connective tissue fragility, can be associated with cardiovascular abnormalities, including aortic root dilatation and multi-valve defects.24 However, this form is obviously different from EDS-PH, since these patients usually present with pronounced craniofacial and skeletal features at birth.
Although we contend that there is little basis for retention of the EDS-PH entity, another overlapping allelic phenotype, X-linked cardiac valvular dystrophy (XCVD), is certainly distinct from the phenotypes studied here. XCVD is characterized by early onset polyvalvular disease in male hemizygotes. All four cardiac valves can be variably involved (dystrophy, prolapse or stenosis) but PH and great artery ectasia have not been reported as features of XCVD.13, 25, 26, 27 It is unknown if joint hyperextensibility is part of this presentation. Missense mutations or intragenic deletions within exons encoding the rod domain of FLNA, constituting presumptive hypomorphic FLNA alleles, underlie this phenotype in males. It is therefore conceivable that this function of FLNA explains the occurrence of cardiac valvular disease in XL-PH. In this study, as in others,9, 11, 12 the cardiovascular phenotype associated with XL-PH is significant and most commonly includes either valvular dystrophy or arterial dilatation. FLNA exhibits pronounced expression within the endothelium, but not the media, of the human aorta suggesting that impairment of an endothelial function rather than a primary structural defect may underlie arterial defects in XL-PH.19
The recognition of progressive vascular defects associated with XL-PH raises the potential for rupture and sudden death in this condition. Descriptions of cohorts of patients with XL-PH do not report such clinical incidents suggesting that this is not a common complication of the disorder,4, 5, 7, 8, 11, 12, 15, 16 although data have not been prospectively sought to answer this specific question. The precise incidence of XL-PH and aortic dilatation is unknown, complicating this uncertainty. In some patients presented here (F1, F2, F5), joint hypermobility and seizures were readily appreciated, but the more significant cardiovascular anomalies were less apparent, thus presenting a case for echocardiographic screening of all individuals with XL-PH to detect vascular complications (specifically aortic/pulmonary dilatation) as well as latent multi-valvular dystrophy. It is notable that some patients with XL-PH can be presented with primarily a joint hypermobility syndrome, while co-morbid cardiovascular manifestations are subclinical. Although skin and joint anomalies are not consistently associated with underlying cardiovascular disease, an awareness and consideration of screening for cardiovascular manifestations is important especially when there is personal or family history of a seizure disorder or X-linked pattern of inheritance of the connective tissue abnormalities.
This cohort study is illustrative of the breadth of expressivity of the XL-PH phenotype but does not address the frequency and proportions of anomalies found within the cutaneous, skeletal and cardiovascular systems in this condition. Further detailed clinical studies in larger cohorts of XL-PH patients are required to better define the correlation between joint laxity in XL-PH patients and vascular involvement, and to formalize the natural history of these complications. In this cohort, only 4 of 10 patients had seizures at the time of presentation, and in some patients, the findings of vascular dilatation and brain heterotopia were incidental discoveries. Future studies will need to address the mode of ascertainment of these patients to provide the best evidence to inform clinical practice.
References
Liu JS : Molecular genetics of neuronal migration disorders. Curr Neurol Neurosci Rep 2011; 11: 171–178.
Andrade DM : Genetic basis in epilepsies caused by malformations of cortical development and in those with structurally normal brain. Hum Genet 2009; 126: 173–193.
Robertson SP, Filamin A : Phenotypic diversity. Curr Opin Genet Dev 2005; 15: 301–307.
Sheen VL, Jansen A, Chen MH et al: Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology 2005; 64: 254–262.
Gómez-Garre P, Seijo M, Gutiérrez-Delicado E et al: Ehlers-Danlos syndrome and periventricular nodular heterotopia in a Spanish family with a single FLNA mutation. J Med Genet 2006; 43: 232–237.
Robertson SP, Twigg SR, Sutherland-Smith AJ et al: Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat Genet 2003; 33: 487–491.
Solé G, Coupry I, Rooryck C et al: Bilateral periventricular nodular heterotopia in France: frequency of mutations in FLNA, phenotypic heterogeneity and spectrum of mutations. J Neurol Neurosurg Psychiatry 2009; 80: 1394–1398.
Parrini E, Ramazzotti A, Dobyns WB et al: Periventricular heterotopia: phenotypic heterogeneity and correlation with FilaminA mutations. Brain 2006; 129: 1892–1906.
de Wit MC, Kros JM, Halley DJ et al: Filamin A mutation, a common cause for periventricular heterotopia, aneurysms and cardiac defects. J Neurol Neurosurg Psychiatry 2009; 80: 426–428.
Sheen VL, Walsh CA : Periventricular heterotopia: new insights into Ehlers-Danlos syndrome. Clin Med Res 2005; 3: 229–233.
de Wit MC, de Coo IF, Lequin MH et al: Combined cardiological and neurological abnormalities due to filamin A gene mutation. Clin Res Cardiol 2011; 100: 45–50.
Lee CH, Wai YY, Wu T : Periventricular nodular heterotopia and cardiovascular defects. Chang Gung Med J 2011; 34: 628–635.
Guerrini R, Mei D, Sisodiya S et al: Germline and mosaic mutations of FLN1 in men with periventricular heterotopia. Neurology 2004; 63: 51–56.
Moro F, Carrozzo R, Veggiotti P et al: Familial periventricular heterotopia: missense and distal truncating mutations of the FLN1 gene. Neurology 2002; 58: 916–921.
Sheen VL, Dixon PH, Fox JW et al: Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum Mol Genet 2001; 10: 1775–1783.
Beighton P, De Paepe A, Steinmann B et al: Ehlers–Danlos Syndromes: Revised Nosology, Villefranche, 1997. Ehlers–Danlos National Foundation (USA) and Ehlers–Danlos SupportGroup (UK). Am J Med Genet 1998; 77: 31–37.
Masurel-Paulet A, Haan E, Thompson EM et al: Lung disease associated with periventricular nodular heterotopia and an FLNA mutation. Eur J Med Genet 2011; 54: 25–28.
de Wit MC, Tiddens HA, de Coo IF et al: Lung disease in FLNA mutation: confirmatory report. Eur J Med Genet 2011; 54: 299–300.
Ferland RJ, Batiz LF, Neal J et al: Disruption of neural progenitors along the ventricular and subventricular zones in periventricular heterotopia. Hum Mol Genet 2009; 18: 497–516.
Pepin M, Schwarze U, Superti-Furga A et al: Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med 2000; 342: 673–680.
Borck G, Beighton P, Wilhelm C et al: Arterial rupture in classic Ehlers-Danlos syndrome with COL5A1 mutation. Am J Med Genet A 2010; 152A: 2090–2093.
Rohrbach M, Vandersteen A, Yiş U et al: Phenotypic variability of the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VIA): clinical, molecular and biochemical delineation. Orphanet J Rare Dis 2011; 6: 46.
McDonnell NB, Gorman BL, Mandel KW et al: Echocardiographic findings in classical and hypermobile Ehlers Danlos syndromes. Am J Med Genet A 2006; 140: 129–136.
Shimizu K, Okamoto N, Miyake N et al: Delineation of dermatan 4-O-sulfotransferase 1 deficient Ehlers-Danlos syndrome: observation of two additional patients and comprehensive review of 20 reported patients. Am J Med Genet A 2011; 155A: 1949–1958.
Bernstein JA, Bernstein D, Hehr U et al: Familial cardiac valvulopathy due to filamin A mutation. Am J Med Genet A 2011; 155A: 2236–2241.
Kyndt F, Gueffet JP, Probst V et al: Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation 2007; 115: 40–49.
Gérard-Blanluet M, Sheen V, Machinis K et al: Bilateral periventricular heterotopias in an X-linked dominant transmission in a family with two affected males. Am J Med Genet A 2006; 140: 1041–1046.
Acknowledgements
The support of the Steven Spielberg Pediatric Research Center, the NIH/NICHD Program Project Grant (HD36657), the Medical Genetics NIH/NIGMS Training Program Grant (5-T32-GM08243), and the Cedars-Sinai General Clinical Research Center Grant (M01-RR00425) for samples collected under CSMC IRB Protocols 0463 and 4232 is acknowledged. SPR is supported by Curekids New Zealand and the Health Research Council of New Zealand. This paper is dedicated to the memory of David L Rimoin.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Reinstein, E., Frentz, S., Morgan, T. et al. Vascular and connective tissue anomalies associated with X-linked periventricular heterotopia due to mutations in Filamin A. Eur J Hum Genet 21, 494–502 (2013). https://doi.org/10.1038/ejhg.2012.209
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.209
Keywords
This article is cited by
-
The phenotypic spectrum of epilepsy associated with periventricular nodular heterotopia
Journal of Neurology (2023)
-
The variants at FLNA and FLNB contribute to the susceptibility of hypertension and stroke with differentially expressed mRNA
The Pharmacogenomics Journal (2021)
-
Cardiovascular and connective tissue disorder features in FLNA-related PVNH patients: progress towards a refined delineation of this syndrome
Orphanet Journal of Rare Diseases (2021)
-
Where the congenital heart disease meets the pulmonary arterial hypertension, FLNA matters: a case report and literature review
BMC Pediatrics (2020)
-
Hemizygous FLNA variant in West syndrome without periventricular nodular heterotopia
Human Genome Variation (2020)
