
Abstract
In the first part, the following mechanisms involved in different forms of cell death are considered, with a view to identifying potential therapeutic targets: tumour necrosis factor receptors (TNFRs) and their engagement by tumour necrosis factor-alpha (TNF-α); poly [ADP-ribose] polymerase (PARP)-1 cleavage; the apoptosis signalling kinase (ASK)-c-Jun N-terminal kinase (JNK) axis; lysosomal permeability; activation of programmed necrotic cell death; oxidative stress, caspase-3 inhibition and parthanatos; activation of inflammasomes by reactive oxygen species and the development of pyroptosis; oxidative stress, calcium dyshomeostasis and iron in the development of lysosomal-mediated necrosis and lysosomal membrane permeability; and oxidative stress, lipid peroxidation, iron dyshomeostasis and ferroptosis. In the second part, there is a consideration of the role of lethal and sub-lethal activation of these pathways in the pathogenesis and pathophysiology of neurodegenerative and neuroprogressive disorders, with particular reference to the TNF-α-TNFR signalling axis; dysregulation of ASK-1-JNK signalling; prolonged or chronic PARP-1 activation; the role of pyroptosis and chronic inflammasome activation; and the roles of lysosomal permeabilisation, necroptosis and ferroptosis. Finally, it is suggested that, in addition to targeting oxidative stress and inflammatory processes generally, neuropsychiatric disorders may respond to therapeutic targeting of TNF-α, PARP-1, the Nod-like receptor NLRP3 inflammasome and the necrosomal molecular switch receptor-interacting protein kinase-3, since their widespread activation can drive and/or exacerbate peripheral inflammation and neuroinflammation even in the absence of cell death. To this end, the use is proposed of a combination of the tetracycline derivative minocycline and N-acetylcysteine as adjunctive treatment for a range of neuropsychiatric disorders.
Similar content being viewed by others

Introduction
Apoptosis and necrosis are two forms of cell death. Apoptosis relies on an intracellular proteolytic cascade which is essentially mediated by two types of caspases (named after the fact that they are proteases with cysteine at the active site and with aspartate targets), namely initiator caspases (such as caspase-8 and caspase-9) and executioner caspases (caspase-3, caspase-6 and caspase-7). Two important mammalian pathways which can activate an initiator caspase are the extrinsic pathway and the intrinsic (or mitochondrial) pathway [1]. Extrinsic apoptosis is activated by the binding to death receptors of members of the tumour necrosis factor (TNF) superfamily of cytokines, such as TNF-α, fibroblast-associated cell surface (Fas) ligand (FasL) and TNF-related apoptosis-inducing ligand (TRAIL). In turn, this induces the formation of the death-induced signalling complex (DISC), which in turn activates caspase-8 (an initiator caspase). Activated caspase-8 activates caspase-3 (an executioner caspase), which executes apoptosis. The activation of the extrinsic apoptotic pathway can lead to the activation of the intrinsic apoptotic pathway, which is dependent on the activity of mitochondria [2], whereby activated caspase-8 cleaves the pro-apoptotic Bcl2 homology (BH) domain BH-3-only protein Bid that induces outer mitochondrial membrane permeabilisation through the interactions of truncated Bid (tBid) with the pro-apoptotic effector Bcl2 family proteins Bax/Bak, resulting in the mitochondrial release of apoptogenic cytochrome c, and other apoptosis inducing factors, such as the second mitochondria-derived activator of caspases/direct inhibitor of apoptosis (IAP)-binding protein (SMAC/DIABLO) [3] into the cytoplasm. Translocation of cytochrome c into the cytosol in turn initiates the assembly of the heptameric apoptosome, containing Apaf-1 and procaspase-9, resulting in the cleavage of the latter and subsequent downstream activation of caspase-3 and apoptotic death [4]. Notably, the intrinsic pathway of apoptosis can be activated following oxidative damage to mitochondrial proteins, DNA damage and peroxidative damage to cardiolipin and other mitochondrial membrane lipids driven by excessive levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS). It is also important to note that TNF-α engagement with TNF receptors (TNFRs) can be anti-apoptotic in certain circumstances and that several mechanisms driving caspase-independent apoptosis also exist, which will be discussed below [5].
Given the acknowledged role of elevated TNF-α, ROS and RNS in the pathogenesis and pathophysiology of neurodegenerative disorders, it is unsurprising that copious evidence exists describing activated or disorganised apoptotic pathways and increased caspase activity in these conditions [6,7,8]. There is also evidence of activated apoptotic programmed cell death pathways in the frontal cortex of patients with major depressive disorder (MDD), and in the anterior cingulate cortex (ACC) and hippocampus of patients with bipolar disorder (BD) and schizophrenia, which are increasingly described as neuroprogressive conditions [9,10,11]. This form of cell death is held to be responsible for the reduced neural and glial density seen in the dorsolateral prefrontal cortex of MDD patients [12] and in the ACC and hippocampus of patients with BD and schizophrenia [13, 14].
Several research teams have demonstrated the existence of ferroptosis, parthanatos, pyroptosis, necroptosis and lysosomal membrane permeability (LMP) as drivers of necrotic cell death in at least some cells and tissues in Parkinson’s disease (PD), Alzheimer’s disease (AD) and other neurodegenerative diseases [15,16,17,18,19]. There is also some evidence implicating lysosomal rupture in the formation of amyloid plaques, especially in AD [20]. This is unsurprising given that high levels of TNF-α, oxidative and nitrosative stress characteristic of neuroprogressive and neurodegenerative conditions (reviewed in references [6, 7, 21]) are known to activate poly [ADP-ribose] polymerase (PARP)-1 and the Nod-like receptor NLRP3 inflammasome, provoke LMP, precipitate necroptosis and exacerbate the development of ferroptosis [22,23,24].
There are numerous studies demonstrating that widespread sub-lethal activation of the TNF-α, PARP-1 and NLRP3 signalling pathways, the presence of lysosomal dysfunction, iron accumulation, lipid peroxidation and downregulation of positive inhibitors of ferroptosis singly and collectively play a causative role in the pathophysiology and pathogenesis of many if not all neurodegenerative diseases [19, 25, 26]. Activation of PARP-1 signalling, the NLRP3 inflammasome, TNF-mediated inflammatory pathways, lysosomal dysfunction, lipid peroxidation and downregulation of positive inhibitors of ferroptosis have also been repeatedly demonstrated in neuroprogressive disorders [10, 27,28,29,30,31,32]. There is also evidence of iron dyshomeostasis in MDD and BD [33, 34], and researchers have previously observed cellular iron accumulation in at least some patients [35, 36]. The situation in schizophrenia, however, is less clear. This is predominantly because of the confounding effects neuroleptics have on the mechanisms of iron homeostasis [37]. Iron accumulation in neurones and microglia of patients with neuroprogressive conditions would not be unexpected however given that elevated levels of oxidative stress and neuroinflammation characteristic of these conditions would be expected to provoke iron dyshomeostasis. Such dysregulation could lead to iron accumulation in microglia and neurones in at least some regions of the brain [38,39,40].
Widespread activation of TNF-α, PARP-1, NLRP3 and the necrosomal molecular switch receptor-interacting protein kinase-3 (RIPK3) in patients with neurodegenerative and neuroprogressive diseases may be of importance from the perspective of pathogenesis and pathophysiology as increased activity of these molecular players can drive and/or exacerbate peripheral inflammation and neuroinflammation even in the absence of cell death [25, 41,42,43]. This may be especially important in neuroprogressive disorders given chronic neuroinflammation—characterised by activated microglia—is an acknowledged source of impaired adult neurogenesis and abnormalities in neurotransmitter systems, which are a recognised source of pathology [29, 44, 45]. Targeting the amelioration of these signalling pathways would seem to be a desirable therapeutic objective in addition to attempting to reduce oxidative stress and inflammation. Accordingly, this review has three objectives: First, to explain the mechanisms involved in the various forms of cell death with a view of identifying therapeutic targets; second, to outline the potential role that sub-lethal and lethal activation of these pathways could play in the pathogenesis and pathophysiology of neurodegenerative and neuroprogressive illnesses; finally, to propose a combination of minocycline and N-acetylcysteine (NAC) as an adjunctive treatment for neuroprogressive conditions focusing on their role in inhibiting molecular players involved in a multitude of cell death pathways.
Apoptosis and necrosis are viewed as opposite extremes on a spectrum of cell death and can occur simultaneously in the same tissue [46, 47]. Necrosis can also take place as a “backup” form of cell death upon failure of caspase-dependent apoptotic mechanisms [48, 49]. This is of importance as levels of TNF-α and ROS play a significant role in determining whether programmed cell death proceeds via apoptosis, whereby excessive levels tend to promote cell death via necrosis while inhibiting apoptosis [46, 47]. A major mechanism driving this preference for necrotic cell death in an environment of prolonged and/or excessive inflammation and oxidative stress is the inhibition of caspase activity and the subsequent collapse of cellular adenosine triphosphate (ATP) generation [50]. This inhibition occurs because the activity of caspases is dependent on the presence of thiol groups in their catalytic sites which are an indispensable element in their capacity to act as proteases and the fact that these groups are exquisitely sensitive to oxidative or nitrosative inactivation in an environment characterised by very high levels of ROS and NO (reviewed by reference [1]). Mechanistically, this phenomenon is underpinned by the inhibition of processes such as proteasome-mediated degradation and gene translation, which consume large amounts of ATP by caspase activation following the instigation of apoptotic machinery [50, 51]. With the inhibition of caspase activity, these processes continue unchecked leading to the collapse of cellular ATP levels which prompts the switch from cell death by apoptosis to cell death by necrosis [50]. One well-documented example of this phenomenon is the inhibition of caspase-8 activity in complex II formed during TNF-α-mediated apoptosis which switches the mode of cell death to TNF-α-mediated necroptosis, while another is the inhibition of caspase-3 which leads to hyperactivation of PARP-1 inducing the development of parthanatos (also known as PARP-1-dependent cell death) [49]. We will now move on to consider these processes in some more detail before discussing other forms of cellular necrosis such as pyroptosis, lysosome-mediated necrosis and ferroptosis.
Mechanisms Underpinning TNF-α- and ROS-Mediated Apoptosis
Engagement of TNFRs by TNF-α
Following stimulation with TNF-α, the TNF receptor TNFR1 translocates into lipid rafts and recruits TNFR-associated protein 2 (TRAF2), the adapter protein TNFR1-associated death domain protein (TRADD), cellular inhibitor of apoptosis protein (cIAP), the E3 ligase linear ubiquitin chain assembly complex (LUBAC) and, finally, receptor-interacting protein kinase-1 (RIPK1) to the plasma membrane, resulting in the formation of a transient signalling platform generally described as complex I [52]. Once in situ, RIPK1 is multiply ubiquitinated and phosphorylated by the E3 ligase LUBAC and cIAP within lipid rafts, leading to rapid activation of nuclear factor-κB (NF-κB) [53, 54] and several downstream anti-apoptotic proteins, such as cellular FLICE (Fas-associated death domain-like IL-1β-converting enzyme)-like inhibitory protein (cFLIP) [52]. Importantly, the ubiquitination status of RIPK1 is an essential element in maintaining NF-κB activation and the stable anchorage of the kinase at the plasma membrane [52, 55]. As such, RIPK1 deubiquitination is mediated by a range of multiple deubiquitinating enzymes (DUBs) such as A20 and cylindromatosis (CYLD), which are upregulated by NF-κB at excessive intracellular concentrations of TNF-α [56, 57]. This inhibits NF-κB signalling, leading to the dissociation of the kinase into the cytoplasm to act as the initial recruiting molecule for the formation of a cytosolic DISC generally described as complex II. This cytosolic signalling platform is comprised of RIPK1, caspase-8, FADD and cFLIP recruited from the cytosol [52, 58]. Here, levels of caspase-8 are deterministic of downstream events. Adequate levels of caspase-8 activate caspase-3 and further downregulate NF-κB signalling by proteolytic cleavage of RIPK1 [59].
Role of PARP-1 Cleavage
Caspase-8 and FADD in tandem inhibit necroptosis by inhibiting the activity of RIPK1 and RIPK3 and CYLD and promote the advent of apoptosis via the cleavage of PARP-1 [58, 60]. Cleavage of PARP-1 by caspase-3 is held to be a universal hallmark of apoptotic cell death [61, 62]. Interestingly, virtually every caspase displays the capacity to inhibit PARP-1, albeit in vitro [63]. Cleavage of PARP-1 by caspase-3 leads to the formation of two catalytic subunits of 89 and 24 kDa with the former exiting into the cytosol and the latter being retained in the nucleus [64, 65] where its binding to DNA inhibits the activity of PARP-1 abrogating DNA repair but conserving levels of ATP, thereby favouring the development of apoptosis [66].
Role of the ASK-JNK Axis
TNF-α-mediated apoptosis is also dependent on ROS production following TNFR engagement [67] and subsequent activation of apoptosis signalling kinase-1 (ASK-1). Briefly, elevated levels of ROS provoke the disengagement of thioredoxin which is attached to ASK-1 in physiological conditions and acts to inhibit its activity [68]. Once activated, this kinase phosphorylates the downstream mitogen-activated protein kinase (MAPK) c-Jun N-terminal kinase-1 (JNK1), which is the effector molecule of an apoptotic cascade [69,70,71]. The sustained apoptotic activity of JNK1 is dependent on TRAF2 and RIPK1 activity and the formation of a TRAF2/RIPK1/JNK1 signalling complex at the plasma membrane [72]. Mechanistically, RIPK1 and TRAF2 are recruited to lipid rafts in an environment of chronic oxidative stress where they associate with JNK1 recruited from the cytoplasm [72,73,74]. Once endocytosed, this signalling complex enables the detrimental effects of JNK on mitochondrial membranes to take place and increases the activity of PARP-1, leading to nicotinamide adenine dinucleotide (NAD+) depletion in the cytosol [74, 75].
Role of Increased Lysosomal Permeability
TNF-α-mediated apoptotic signalling triggers lysosomal permeabilisation and the subsequent release of lysosomal proteinases known as cathepsins into the cytoplasm; this involvement of cysteine-dependent cathepsins in the cytoplasm secondary to the development of lysosomal permeabilisation plays a significant role in the development of apoptosis [76,77,78]. Some debate remains as to the major mechanisms underpinning the pro-apoptotic role of cathepsins, but the weight of evidence suggests that their role in caspase-dependent apoptosis may be limited to caspase activation while cathepsins B, L and D would appear to be pivotal players in instigating or exacerbating caspase-independent cell death in the brain [79]. Some cathepsins, such as cathepsin D, have the capacity to induce mitochondrial membrane permeabilisation and mitochondrial permeability transition via proteolytic activation of the Bcl-2 family members Bid, Bax and Bak. This results in Bax/Bak activation, the formation of pores in the outer mitochondrial membrane and the escape of cytochrome c, apoptosis-inducing factor (AIF) and SMAC/DIABLO from the inter-membrane space into the cytoplasm triggering apoptotic death [80,81,82].
Mechanisms Underpinning Non-apoptotic Programmed Cell Death
Increased Levels of ROS and TNF-α and the Activation of Programmed Necrotic Cell Death
In an environment where caspase-8 activity (in complex II, formed following TNF ligation of TNFR; as described above) is inhibited by cIAP, increased oxidative stress or post-translational modifications [1], complex II is unable to initiate intrinsic apoptosis or inhibit the activity of RIPK1 and RIPK3 [83]. In such circumstances, RIPK1 and RIPK3 autophosphorylate and transphosphorylate with one another to form a complex described as the necrosome. In addition to containing RIPK1 and RIPK3, this cytosolic structure also includes cIAP and the mixed lineage kinase domain-like protein (MLKL) [84, 85]. The conditional DUBs CYLD and A20 deubiquitinate RIPK1 [52, 86, 87]. RIPK1 subsequently phosphorylates RIPK3, which in turn phosphorylates MLKL leading to its oligomerisation [87, 88]. This change in MLKL conformation triggers recruitment to the plasma membrane. Once in situ, it obtains anchorage by binding negatively charged phosphatidylinositol phosphate (PIP), before translocating into lipid rafts. Here, it induces the type of necrosis known as necroptosis, by triggering Na+ and Ca2+ influx into the cell [87].
Considerable evidence now exists suggesting that ROS have a direct role in driving necroptosis and apoptosis via routes which are independent of caspase activation [89]. For example, several research teams have reported that excessive levels of TNF can induce a large increase in mitochondrial ROS production which enhances necrosome formation (see, for example, reference [90]). Indeed, recent evidence suggests that physiologically elevated levels of mitochondrial ROS production are an essential element in RIPK3 recruitment into the necrosome [91]. The indispensable role ROS play in enabling or driving TNF-α-induced apoptosis or necroptosis is further emphasised by data demonstrating that these forms of cell death are inhibited by the administration of free radical scavengers even in an environment where extracellular and intracellular TNF-α levels are excessive [92, 93] (review by reference [91]).
Increased Oxidative Stress, Caspase-3 Inhibition and Development of Parthanatos
Increased levels of oxidative stress and the subsequent inhibition of caspase-3 lead to persistent PARP-1 hyperactivation. This promotes excessive consumption of NAD+ and the exhaustion of cellular ATP, leading to necrotic cell death [94,95,96]. Mechanistically, this form of caspase-independent programmed necrosis involves the sequential activation of PARP-1, calpains, Bid- and Bax-induced mitochondrial membrane depolarisation, mitochondrial permeability transition pore opening and efflux of AIF into the cytoplasm and ultimately the nucleus [97, 98]. Once in the nucleus, AIF associates with the phosphorylated histone H2AX to form a DNA-degrading complex that induces chromatinolysis and cell death by parthanatos [99]. Importantly, while direct depletion of NAD+ by PARP-1 is responsible for a catastrophic decline in ATP generation and efflux of AIF is mediated by calpain, the initial signalling of PARP-1 to mitochondria is mediated by the downstream activation of JNK1 [100] (review by reference [101]).
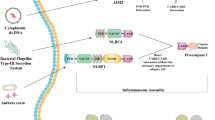
Activation of Inflammasomes by ROS and the Development of Pyroptosis
ROS can activate the complex cytosolic multiprotein signalling platforms known as inflammasomes [23, 102]. Typically, these structures are composed of sensor and adaptor proteins and the zymogen procaspase-1, with the latter processed into active caspase-1 following inflammasome complex assembly [103]. Caspase-1 in turn cleaves the zymogen forms of interleukins IL-1β and IL-18 leading to their activation and the development or exacerbation of inflammation and in many cases pyroptotic cell death [103, 104]. Inflammasome activation can also lead to pyroptotic cell death via caspase-1- and caspase-11-mediated cleavage of the cytoplasmic protein gasdermin D (Gsdmd) and other cell death mediators whose actions are not currently understood [105, 106]. Pyroptosis induces rapid plasma membrane rupture and excessive release of pro-inflammatory cytokines (PICs) and chemokines which may aggravate inflammation-mediated neuronal death [107, 108]. These molecular players are also mediators of activated leucocyte recruitment from the periphery which exacerbate inflammatory responses causing severe central nervous system (CNS) tissue damage in neuropathological conditions. As these factors have been shown to mediate the recruitment of other immune cells from the peripheral circulation, an abundance of leukocytes is attracted to the inflammation sites, and subsequent inflammatory responses can cause severe tissue damage in the CNS under neuropathological conditions [109].
In the case of activation of the inflammasomes NLRP3 and absent in melamoma-2 (AIM-2) however, the execution of this form of lytic and inflammatory cell death is not inevitable, as mechanisms exist that can inhibit the signalling of and accelerate the clearance of these complexes [110]. Notably, chronic low level activation of NLRP3 plays a major causative role in the pathogenesis and pathophysiology of a range of autoimmune, metabolic, neurological and neuroprogressive illnesses (review by references [111, 112]). The assembly and performance of inflammasomes is regulated by an almost bewildering array of enzymes, including, but not limited to inhibitory kappa-B kinase-1 (IKK1), IKK2, LUBAK and JAK1 (see reference [110]). Importantly, given the levels of nitrosative and oxidative stress seen in patients with neuroprogressive disorders, inflammasome activity can also be regulated by levels of nitric oxide-induced protein S-nitrosylation [113]. This is also true of caspase-1 activity, which may be inactivated in such a cellular environment [114] and which may also partly explain how NLRP3 activation in these circumstances may have a range of sub-lethal consequences in at least some regions and tissues in the brain. While oxidative stress clearly plays a role in inflammasome activation, high levels of ROS and NOS and/or the development of calcium dyshomeostasis can also induce LMP in some circumstances, leading to another “explosive” form of cell death described as lysosomal-mediated necrosis (LMN), described in the following section.
Oxidative Stress, Calcium Dyshomeostasis and Iron in the Development of LMN and LMP
LMN is another form of cell death largely mediated by the lethal proteolytic effects of cysteine cathepsins entering the cytosol following complete lysosomal membrane rupture [115,116,117]. This form of cell death is characterised by the proteolysis of several crucial inflammatory protein zymogens, such as caspase-1 and IL-1, and severe damage to the cell plasma membrane via indeterminate mechanisms [117]. Lysosomal rupture may also occur as an upstream event in at least some forms of apoptosis [118, 119]. The weight of evidence would suggest that the degree and perhaps the speed of lysosomal rupture may be decisive with moderate lysosomal rupture inducing apoptosis, while pronounced lysosomal leakage results in necrosis devoid of caspase activation [120]. The mechanisms underpinning this dichotomy are not fully understood, but accumulating evidence suggests that the massive influx of low molecular mass iron into the cytosol secondary to complete lysosomal rupture results in the inactivation of functional cysteine thiol groups in the catalytic site of procaspase-9, thereby preventing its activation [121, 122].
Several members of the cathepsin family are involved in mediating LMN depending on the nature of cytotoxic stimuli involved. For example, cathepsins S and B enable alum (aluminium hydroxide)-mediated necrosis and the development of adaptive immunity following immunisation [123,124,125], while cathepsin D mediates LMN following lysosomal permeability driven by high levels of intralysosomal iron [126]. Given the redox-active nature of intralysosomal iron ions, and their ability to engage in the Fenton reaction with hydrogen peroxide, it is unsurprising that rapid release of intralysosomal iron into the cytosol following lysosomal rupture is a significant driver of cell death, either alone or in synergy with other oxidative drivers of DNA damage and LMP [127,128,129]. Increased iron accumulation within cellular lysosomes and in other cellular compartments is held to be a major contributing factor in ferroptosis. In this context, it is noteworthy that high levels of ROS, RNS and TNF-α are key drivers of iron dyshomeostasis, which consequentially leads to intracellular iron accumulation [39, 130, 131]. There are many factors involved in the development of ferroptosis outside of iron dyshomeostasis however, and this form of cell death appears to be unique in that genetic susceptibility seems to have a major influence. We will consider this element, as well as the other factors driving ferroptotic cell death in the next section.
Oxidative Stress, Lipid Peroxidation, Iron Dyshomeostasis and Ferroptosis
Ferroptosis is characterised by the accumulation of iron and lipid hydroperoxides and their metabolites in the cytosol and is effected by fatal peroxidation of polyunsaturated fatty acids (PUFAs) in the plasma membrane [132,133,134,135]. There is considerable evidence implicating disturbed iron homeostasis in ferroptosis; for example, impaired iron regulatory protein 2 (IRP2) activity coupled with unusually high levels of the transferrin receptor, transferrin and mitochondrial ferritin has been linked with this process [88, 132, 136]. Some other elements which appear to be peculiar to the process of ferroptosis include the need for active lysosomes [137] as well as the involvement of mitochondrial glutaminase-2 and subsequent glutaminolysis [26, 132, 138]. In addition, several mitochondrial genes are associated with the development of this form of cell death, and there is evidence suggesting peroxidation of cardiolipin [26, 139]. Despite this, mitochondrial ROS, calcium dyshomeostasis and the interaction of truncated Bid, Bax and Bad do not appear to be triggers of ferroptosis [24, 140].
Recent evidence also indicates that the increased activity of certain types of lipoxygenase (LOX), most notably 15-LOX, and subsequent oxidation of arachidonic acid and phosphatidylethanol are indispensable elements in the induction of ferroptotic cell death [134, 141, 142]. Ferroptosis is negatively regulated by nuclear factor (erythroid-derived 2)-like 2 (Nrf2 or NFE2L2 or NF-E2-related factor 2), glutathione, glutathione peroxidase-4 and the glutamate/cystine antiporter system and positively regulated by NADPH oxygenase and p53 [26]. Lipoxygenase-mediated peroxidation of PUFAs, with the resultant production of oxidised phosphatidylethanolamine and the two fatty acyls, arachidonoyl and adrenoyl, coupled with the mediation by the acyl-CoA synthetase long-chain family member ACSL4 of the production of 5-hydroxyeicosatetraenoic acid, would appear to be the ultimate executioners of ferroptosis [134, 142, 143]. It would also appear that the lipid composition of the lipid membrane is an important factor, with an increased concentration of long-chain omega-6 PUFAs conveying particular risk [144]. This is important given evidence now suggests that the level of ACSL4 might well dictate the sensitivity towards the development of ferroptosis by influencing the lipid composition of the cell membrane [144]. Individual differences in the activity of ACSL4 might in part explain the strong genetic component in the sensitivity to ferroptosis, and manipulation of this enzyme may ultimately offer an appropriate therapeutic intervention [141, 144].
The role of iron in this process remains a matter of debate [145]; however, iron dyshomeostasis would appear to be involved, and there is evidence that ferroptosis includes ferritinophagy and subsequent release of redox-active iron into the cytosol via the nuclear receptor co-activator 4 (NCOA4)-regulated autophagy pathway [26, 143]. There is also some evidence that iron-mediated activation of phosphorylase kinase G2 (PHKG2) directly mediates lipoxygenase-mediated peroxidation of PUFAs leading to the accumulation of lethal hydroperoxides, but this datum is yet to be replicated [134]. Finally, a recent study conducted by Muller and colleagues produced data suggesting that ferroptosis and necroptosis are two alternative cell death pathways and inhibition of one provokes induction of the other [146]. The mechanisms underpinning this phenomenon are yet to be delineated however.
Having examined the mechanisms involved in cell death mediated by TNF-α, ROS and RNS, we now turn our attention to the second objective of the paper, namely an examination of the potential roles of these cell death pathways both at lethal and sub-lethal levels of activation in the pathogenesis and/or physiology of neuroprogressive and neurodegenerative conditions.
Putative Involvement of Cell Death Machinery in the Pathogenesis and Pathophysiology of Neurodegenerative and Neuroprogressive Illnesses
The TNF-α-TNFR Signalling Axis
High levels of TNF-α are found in the brains and cerebrospinal fluid (CSF) of patients with PD and AD and are implicated in the pathogenesis and pathophysiology of both illnesses [147,148,149,150]. Consistent with this, increases in TNFR1 activity and decreases in TNFR2 activity have also been frequently reported in both diseases [150,151,152]. There would appear to be a difference in the distribution of this abnormal brain TNFR activity in these two conditions however, with this phenomenon being widespread in AD patients but confined to the substantia nigra in patients diagnosed with PD [152]. The importance of increased TNF-α and TNFR1 activity in the pathogenesis of AD is highlighted by data suggesting that levels of TNFR1 activity are predictive of the transition between mild cognitive impairment and AD [153]. It is also interesting to note that TNFR1 activity is predictive of the development of neurocognitive disturbance in PD, but there would appear to be no published research investigating TNFR1 activity and the severity of motor symptoms [154]. There would also appear to be a dearth of data investigating TNFR activity in other chronic neurodegenerative diseases, although levels of TNFR1 and TNFR2 activity are abnormal in the animal model of multiple sclerosis (MS), described as experimental autoimmune encephalomyelitis [152].
Elevated TNF-α levels and TNFR1 activity are also seen in the brain and periphery in MDD, and the connection between TNF signalling and illness progression and severity is well documented [155, 156]. Interestingly, while TNF-α levels are elevated in all phases of BD, TNFR activity would appear to differ from that observed in MDD with elevations in the levels of TNFR1 and TNFR2 activity being a replicated finding even in patients during the euthymic phase of the illness [157]. Unsurprisingly, TNFR1 activity is also elevated in patients who have received a diagnosis of schizophrenia [158], and there are data demonstrating that increased levels of TNF-α are associated with acute exacerbations of this illness [159].
Dysregulation of ASK-1-JNK Signalling
Dysregulated JNK signalling is implicated in the pathophysiology of PD [160] and AD [161], by facilitating dopaminergic neuronal death and modulating the activity of p53 upregulated modulator of apoptosis (PUMA), respectively [162]. As well as regulating neuronal apoptosis, JNKs also regulate brain morphogenesis and the architecture of dendrites during neural development and govern crucial neurone-specific activities such as the formation of long-term memory and synaptic plasticity (review by reference [163]). Data implicating JNK1 in the regulation of dendrite arborisation in the cerebellum [164] and hippocampus [165] are of particular interest as overgrowth of dendrites during development is associated with the pathogenesis of schizophrenia and autism spectrum disorders [166]. Moreover, there is direct evidence of JNK1 signalling abnormalities in both conditions [167, 168]. For example, genetic risk for schizophrenia is associated with the JNK pathway [168], and the activity of JNK1 in the cerebral cortex is heavily reliant on a kinase with genetic locus 16p11.2, a well-documented genetic susceptibility locus for schizophrenia and indeed autism [167, 169]. Furthermore, the interleukin-1 receptor accessory protein like-1 gene, implicated in autism, transduces signals via JNK activation [170]. It is also worthy of note that loss of function of some JNK family members owing to chromosomal translocations is associated with the development of intellectual disability [166, 171].
ASK-1 is activated by oxidative stress, nitrosative stress, endoplasmic reticulum (ER) stress, PICs, lipopolysaccharide (LPS) and Ca2+ influx [172] and plays a pivotal role in cell differentiation and the development of chronic inflammation as well as having a well-documented role in driving apoptosis [173]. This kinase acts as a major vehicle for the direct transduction of ROS signalling to downstream targets, and its level of activity influences both the rate of progression and the severity of several neurodegenerative diseases [174, 175]. For example, an amyloid beta-mediated increase in ASK-1-JNK1 activity appears to be an important element driving neuronal death in AD [176], while ASK-1-JNK1-mediated dopaminergic neuronal death appears to be involved in the pathogenesis of PD [177]. More generally, ASK-1 activation plays a significant role in driving the pathophysiology of numerous diseases involving grossly dysfunctional cellular responses to the advent of ER stress and oxidative stress [178].
Prolonged or Chronic PARP-1 Activation
Unsurprisingly, several research teams have adduced evidence of caspase-3-mediated cleavage of PARP-1 in a number of neurological conditions such as MS and AD, and the process is also implicated in the development of N-methyl-d-aspartate (NMDA) receptor-mediated excitotoxicity (review by reference [179]). PARP-1 mediates the transfer of poly-adenosine diphosphate (poly-ADP) from NAD+ to DNA thereby provoking chromatin remodelling and changes in DNA methylation and histone acetylation and thus acting as an epigenetic regulator of gene transcription. Furthermore, PARP-1 also mediates the transient attachment of poly-ADP to target proteins thereby acting as a post-translational regulator of protein function [180, 181]. From the perspective of this paper, it is of particular interest that PARP-1 activity regulates the activation levels and differentiation patterns of T and B lymphocytes, via the regulation of transcription factors such as nuclear factor of activated T cells (NFAT) and the activity of inflammatory pathways in response to acute or chronic cellular stressors [182]. In particular, increased PARP-1 activity sustains the production and activity of PICs such as TNF-α and IL-1β, chemokines such as macrophage inflammatory protein (MIP)-2 (also known as chemokine (C-X-C motif) ligand 2 (CXCL2)), and selectins in the periphery and in glial cells of the brain [183].
Apart from its well-documented role in DNA damage repair, PARP-1 modulates many processes in glial cells contributing to the development of neuroinflammation [184, 185]. For example, PARP-1 is an indispensable binding partner in the NF-κB-mediated activation of microglia and in enabling the transcription of inflammatory molecules such as IL-1β, TNF-α and NO [184, 186]. PARP-1 also regulates the activation of astrocytes and their production of inflammatory cytokines and chemokines [185]. Furthermore, the activation of PARP-1 in astrocytes leads to profound bioenergetic depletion in these glia and subsequent inhibition of glutamate reuptake, thereby contributing to the development of NMDA receptor excitotoxicity, which is a feature of neuroprogressive diseases [187]. PARP-1 also exerts a number of physiological roles in the CNS and its levels are regulated by neural activity [188, 189]. The weight of evidence suggests that this transcription factor plays an important role in the consolidation and reconsolidation of long-term memory and is particularly involved in the extinction of contextual fear memory [190].
Role of Pyroptosis and Chronic Inflammasome Activation
Pyroptosis has been observed in microglia, astrocytes and neurones [191, 192]. Moreover, several authors have reported the involvement of abnormal levels of NLRP3 signalling in a wide range of neurological disorders [193,194,195]. For example, increased activity of NLRP3 and caspase-1 in the brains of AD patients has been repeatedly reported [194, 196]. Similarly, other research teams have reported increases in the expression and activity of NLRP3, IL-1β, IL-18 and caspase-1 in plaques of MS patients and the tissues of amyotrophic lateral sclerosis (ALS) (or motor neurone disease) patients [195, 197,198,199].
Increased levels and activity of caspase-1, IL-1β and IL-18 have also been recorded in several disorders characterised by the existence of chronic neuroinflammation [200,201,202]. Importantly, from the perspective of generating pathology, both IL-1β and IL-18 bind to their respective cognate receptors on neurones, microglia and astrocytes, thereby triggering a highly complex pattern of inflammatory signalling pathways culminating in increased transcription of pro-inflammatory genes [19]. The activation of these pathways and increased transcription of these genes are also associated with the development of cognitive decline and the development of long-term neuroprogressive illnesses [203]. Additionally, there is evidence suggesting that NLRP3 activation in microglia is a driver of neuroinflammation in MDD [28, 204] and a source of chronic immune activation and mitochondrial complex I dysfunction seen in the brains of patients with BD and schizophrenia [205, 206]. Finally, it should be emphasised that NLRP3 activation is increasingly recognised as being a causative factor in the inflammation, mitochondrial dysfunction and chronic oxidative stress seen in autoimmune and autoinflammatory diseases (review by reference [207]).
Role of Lysosomal Permeabilisation
LMN is another form of cell death largely mediated by the lethal proteolytic effects of cysteine cathepsins entering the cytosol following the development of lysosomal permeabilisation [115,116,117]. LMN is characterised by proteolysis of several crucial (pro-)inflammatory zymogens such as caspase-1 and IL-1 and severe damage to the cell plasma membrane via unclear mechanisms [117]. Several members of the cathepsin family are involved in mediating LMP depending on the nature of the cytotoxic stimuli involved. For example, cathepsins S and B enable alum-mediated necrosis and the development of adaptive immunity following immunisation [123,124,125]. In the absence of such external stimuli, however, lysosomal permeabilisation is mediated by high levels of oxidative stress, increased activity of calpains and unusually high lysosomal iron content [126, 208, 209]. Recent evidence also indicates that LMP in vivo is dependent on the activity of the protein signal transducers and activators of transcription-3 (Stat3) [210]. The importance of intralysosomal iron load in this form of cell death is emphasised by data revealing that cathepsin D mediates cell death following LMP driven by high levels of intralysosomal iron [126]. This is consistent with the weight of evidence indicating that that rapid release of intralysosomal iron into the cytosol following such lysosomal rupture is a major driver of cell death either alone or in synergy with other oxidative drivers of DNA damage and lipid membrane permeabilisation [128, 211, 212].
LMP is a potentially lethal event because the ectopic presence of lysosomal proteases in the cytosol causes the digestion of vital proteins and the activation of additional hydrolases including caspases. The latter process is usually mediated indirectly, through a cascade in which LMP causes the proteolytic activation of Bid (which is cleaved by the two lysosomal cathepsins B and D). Bid activation then induces mitochondrial outer membrane permeabilisation, resulting in cytochrome c release and apoptosome-dependent caspase activation. However, massive LMP often results in cell death without caspase activation; this cell death may adopt a subapoptotic or necrotic appearance [209].
Perhaps the strongest evidence of the involvement of frank lysosomal rupture in the pathogenesis of a neurodegenerative disease has been provided by the finding that lysosomal dysfunction is associated with AD and that lysosomal dysfunction is also associated with both the formation of β-amyloid peptide (Aβ) and the hyperphosphorylation of tau protein; two of the most important neuropathological features of AD are amyloid plaques and neurofibrillary tangles, which are caused by dysfunction and accumulation of Aβ and abnormally phosphorylated tau, respectively (review by reference [213]). There is also some evidence to support the view that lysosomal rupture induced by α-synuclein is a cause of dopaminergic neuronal death in PD [214]. More generally, LMP is increasingly regarded as a major driver of ROS-mediated cell death [209, 215], and calpain-mediated LMP is considered to be a major element in the development of pathological lysosomal dysfunction and neuronal necrosis characteristic of most, if not all, neurodegenerative diseases [208, 216, 217] (review by reference [218]). However, there would appear to be no published data investigating the potential existence of LMP as a source of pathology in neuroprogressive disorders despite the presence of chronic oxidative stress, increased calpain activity and lysosomal dysfunction [219, 220].
Role of Necroptosis
There is widespread evidence of necroptosis in a broad array of neurological, neuroprogressive, autoimmune and other inflammatory diseases, as this process is the source of extracellular damage-associated molecular patterns (DAMPs) such as high-mobility group box 1 (HMGB1), S100B and mitochondrial DNA seen in all these conditions [51, 221, 222]. The production of DAMPs is of prime importance as these molecules activate pathogen recognition receptors on antigen-presenting cells, leading to the chronic activation of immune and inflammatory pathways. DAMP production therefore is a major driver of the chronic inflammation, immune activation and oxidative stress seen in illnesses ranging from AD, to systemic lupus erythematosus (SLE) to MDD [223, 224]. While DAMPs play a role in the pathogenesis and pathophysiology of an extensive array of medical conditions (review by references [225, 226]), the role extracellular mitochondrial DNA plays in initiating and maintaining what has been described as the “neuroinflammation-neurodegeneration alliance”—which drives the progression of neurodegenerative diseases—is particularly pertinent for the matters considered in this paper [227, 228]. DAMPs such as mitochondrial DNA, S100B and the 70-kDa heat shock proteins (HSP70s) are also a major cause of chronic immune activation in BD [229], schizophrenia [230] and MDD [45, 223]. Defective caspase-8 activation and increased levels RIPK1, RIPK3 and MLKL have been demonstrated in the cortical lesions of MS patients post-mortem [231] and would appear to be the major driver of exocytotic neuronal death, particularly as far as hippocampal neurones are concerned [232].
The Role of Ferroptosis
Rapidly accumulating data suggest that ferroptosis is an important mediator of cell death in PD and AD [233,234,235]. Furthermore, there is mounting evidence that the processes underpinning this form of cell death, such as decreased glutathione and glutathione peroxidase-4 (GPx-4) levels, are involved in the pathogenesis and pathophysiology of BD [236, 237], schizophrenia [237,238,239] and MDD [237, 240] (review by reference [241]). Readers interested in the classification of lipid endoperoxides and hydroperoxides and the mechanisms underpinning their production and toxic effects such as inducing lipid membrane permeabilisation, producing toxic aldehydes and acting as precursor molecules for inflammatory prostaglandins are invited to consult excellent reviews on the subject by references [242, 243].
Minocycline and NAC as Therapeutic Inhibitors of Cell Death Machinery
Minocycline
The capacity for minocycline to provide neuroprotection and ameliorate neuroinflammation in vivo has been established by several research teams utilising various animal models of neurodegenerative conditions, such as AD [244], PD [245], ALS [246], Huntington’s disease [247] and MS [248, 249] (review by reference [250]). Putative mechanisms underpinning these effects include the inhibition of microglial activation and proliferation [251, 252]. Unsurprisingly, there are also considerable data demonstrating that minocycline also suppresses microglial production of IL-1β, IL-6, TNF, NADPH oxidase and inducible nitric oxide synthase (iNOS), as well as inhibiting T cell egress into the brain [246, 253,254,255]. The mechanisms whereby minocycline therapy reduces microglial activity and neuroinflammation remain to be fully elucidated, but it would appear that inhibition of p38/MAPK and metalloproteinase-9 plays a pivotal role [251, 252, 256].
Minocycline inhibits apoptotic and necrotic cell death in vivo via a range of different mechanisms including the direct inhibition of caspase-1 and caspase-3 in the cytosol [247, 257]. Many of the anti-apoptotic effects of this tetracycline derivative occur at the level of the mitochondria. One important example is the prevention by minocycline of Ca2+ uptake into mitochondria, thereby preventing the development of permeability transition and the collapse of the transmembrane potential difference. This resultantly prevents the release of pro-apoptotic molecules such as cytochrome c, SMAC/DIABLO and AIF into the cytosol [258,259,260]. Minocycline also modulates levels of Bcl2 [261, 262], normalises the Bax/Bcl2 ratio [263] and also inhibits the activity of Bid, thereby preventing the downstream activation of caspases 3, 8 and 9 [8, 264,265,266]. It should be noted, however, that these cytoprotective effects of minocycline may be replaced by toxic effects when cells are exposed to low doses of minocycline (around 50 to 100 μM), which occur at the expense of impaired mitochondrial function and decreased ATP production at these lower concentrations, owing to minocycline-induced reductions in levels of cytochrome c and NAD+ and in activity of enzymes of the electron transport chain [267, 268].
There is also in vivo evidence suggesting that minocycline may have the capacity to mitigate pyroptosis via direct inhibition of PARP-1 activity [266, 269] as well as the ability to chelate Ca2+, which may suppress the activation of several, if not all, members of the calpain family [270,271,272]. Additionally, Shahzad and colleagues recently reported that minocycline stabilises endogenous Nrf2 by reducing its levels of ubiquitination leading to the inhibition of an NLRP3-inflammasome-induced rodent model of diabetic nephropathy. This is of interest given the acknowledged role of Nrf2 inhibition in the development of ferroptosis [24, 273]. In this context, it is also worth noting that the capacity for minocycline to inhibit the development of lipid peroxidation in vivo has also been established in several studies [274, 275]. Several research teams have also adduced evidence demonstrating that minocycline attenuates iron overload following experimental induction of intracerebral haemorrhage in rodents [138, 276, 277]. This reduction in cellular iron levels was accompanied by lowered levels of haem oxygenase-1, transferrin and non-haem iron in the brain, and ferritin in the systemic circulation. Importantly, the reduction in iron load also led to a significant reduction in objective measures of CNS damage and a significant increase in the integrity of the blood-brain barrier [138, 276, 277].
Rodent studies indicate that 50 mg/kg of minocycline has the capacity to reduce the anhedonia and sickness behaviour secondary to LPS-induced microglial activation and subsequent neuroinflammation via the reduction in levels of inflammatory mediators and in indolamine-2,3-dioxygenase (IDO) [253, 278]. This is of particular interest given the results of a recent human trial investigating the therapeutic potential of 200 mg daily minocycline for 3 months as an adjunctive treatment for MDD, which reported significant improvements in a range of clinical parameters although not in the primary endpoint [279]. These results are very similar to, although somewhat better than, those obtained by the use of NAC as an adjunctive treatment for MDD in an earlier study [280]. This may be particularly pertinent given evidence that co-administration of NAC and minocycline had synergistic effects on the attenuation of neuroinflammation in a rodent model of traumatic brain injury, superior to the either preparation alone [281]. This area appears to be worthy of further investigation and may offer a way forward where the properties unique to each molecule may provide the basis for combination therapy for MDD and possibly other neuroprogressive disorders. Accordingly, we will now discuss the possible contribution of NAC in reducing levels of ROS and TNF-α and in inhibiting various molecular players and pathways underpinning the machinery of cell death.
NAC
Numerous researchers investigating the use of NAC supplementation in animal and human studies have published findings affirming the anti-inflammatory, antioxidant and cytoprotective properties of NAC in vivo [282,283,284]. These findings include increased glutathione, reduced levels of ROS as evidenced by decreased levels of hydrogen peroxide and hydroxyl radicals, reduced levels of lipid peroxidation evidenced by reduced levels of malondialdehyde and 4-hydroxy-2-trans-nonenal, together with restored calcium homeostasis and decreased calcium ion entry into mitochondria [285,286,287,288]. Such supplementation also leads to improved mitochondrial performance as evidenced by increased ATP production, increased mitochondrial membrane potential difference and increased outer mitochondrial membrane stability [286,287,288]. Further evidence of the in vivo efficacy of NAC was provided by Kose and Naziroglu [289], who reported that NAC supplementation in polycystic ovary syndrome patients was associated with reduced levels of lipid peroxidation, ROS, mitochondrial membrane depolarisation, caspase-9 and caspase-3, and increased levels of glutathione and GPx [289]. Other research teams have reported that in vivo NAC supplementation increases levels of cFLIP and cIAP and reduces levels of Bax while increasing levels of Bcl2 and decreasing translocation of cytochrome c and AIF into the cytoplasm, hence inhibiting many processes driving intrinsic apoptosis [286].
It would also appear that many of the biochemical consequences and symptomatic improvements produced by NAC supplementation occur via increases in the activity of the oxidative stress-inducible cystine/glutamate exchange system (system Xc −) rather than merely serving as a precursor molecule providing the cysteine needed to enable increased synthesis of glutathione [290,291,292]. The mechanisms underpinning the NAC-induced increases in the activity of system Xc − would appear to be relatively complex and involve the stimulation of as yet undelineated cellular signalling pathways [292,293,294]. Predictably, the profound anti-inflammatory and antioxidant capacity of NAC has made the molecule the subject of intense research in the fields of neurology and neuropsychiatry; reviews of trials in neurology and psychiatry are given by references [295, 296].
Conclusions
Various forms of cell death are involved in the pathogenesis and pathology of a wide range of neuropsychiatric disorders. Related signalling pathways, in addition to oxidative stress and generalised inflammatory processes, appear to offer good therapeutic targets. In particular, a combination of minocycline and NAC may offer a relatively safe and tolerable form of adjunctive treatment for such disorders, which currently can be difficult to treat.
Authorships
All authors contributed equally to the construction and editing of the paper.
References
Circu ML, Aw TY (2010) Reactive oxygen species, cellular redox systems and apoptosis. Free Radic Biol Med 48(6):749–762. https://doi.org/10.1016/j.freeradbiomed.2009.12.022
Han D, Ybanez MD, Ahmadi S, Yeh K, Kaplowitz N (2009) Redox regulation of tumor necrosis factor signaling. Antioxid Redox Signal 11(9):2245–2263. https://doi.org/10.1089/ars.2009.2611
Nguyen KC, Willmore WG, Tayabali AF (2013) Cadmium telluride quantum dots cause oxidative stress leading to extrinsic and intrinsic apoptosis in hepatocellular carcinoma HepG2 cells. Toxicology 306:114–123. https://doi.org/10.1016/j.tox.2013.02.010
Kupsco A, Schlenk D (2016) Molecular mechanisms of selenium-induced spinal deformities in fish. Aquat Toxicol 179:143–150. https://doi.org/10.1016/j.aquatox.2016.09.001
Sinha K, Das J, Pal PB, Sil PC (2013) Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol 87(7):1157–1180. https://doi.org/10.1007/s00204-013-1034-4
Morris G, Walder K, Puri BK, Berk M, Maes M (2015) The deleterious effects of oxidative and nitrosative stress on palmitoylation, membrane lipid rafts and lipid-based cellular signalling: new drug targets in neuroimmune disorders. Mol Neurobiol 53:4638–4658. https://doi.org/10.1007/s12035-015-9392-y
Morris G, Berk M (2015) The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med 13(68). doi:https://doi.org/10.1186/s12916-015-0310-y
Friedlander RM (2003) Apoptosis and caspases in neurodegenerative diseases. N Engl J Med 348(14):1365–1375. https://doi.org/10.1056/NEJMra022366
Shelton RC, Claiborne J, Sidoryk-Wegrzynowicz M, Reddy R, Aschner M, Lewis DA, Mirnics K (2011) Altered expression of genes involved in inflammation and apoptosis in frontal cortex in major depression. Mol Psychiatry 16(7):751–762
Benes FM, Matzilevich D, Burke RE, Walsh J (2005) The expression of proapoptosis genes is increased in bipolar disorder, but not in schizophrenia. Mol Psychiatry 11(3):241–251. https://doi.org/10.1038/sj.mp.4001758
Boyajyan AS, Chavushyan AS, Zakharyan RV, Mkrtchyan GM (2013) Markers of apoptotic dysfunctions in schizophrenia. Mol Biol 47(4):587–591. https://doi.org/10.1134/s002689331304002x
Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP (2002) Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb Cortex 12(4):386–394
Benes FM, Vincent SL, Todtenkopf M (2001) The density of pyramidal and nonpyramidal neurons in anterior cingulate cortex of schizophrenic and bipolar subjects. Biol Psychiatry 50(6):395–406
Ongur D, Drevets WC, Price JL (1998) Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci U S A 95(22):13290–13295
Chen D, Eyupoglu IY, Savaskan N (2017) Ferroptosis and cell death analysis by flow cytometry. In: Gilbert DF, Friedrich O (eds) Cell viability assays: methods and protocols. Springer, New York, pp. 71–77. https://doi.org/10.1007/978-1-4939-6960-9_6
Venderova K, Park DS (2012) Programmed cell death in Parkinson’s disease. Cold Spring Harb Perspect Med 2(8):a009365. https://doi.org/10.1101/cshperspect.a009365
Burguillos MA, Hajji N, Englund E, Persson A, Cenci AM, Machado A, Cano J, Joseph B et al (2011) Apoptosis-inducing factor mediates dopaminergic cell death in response to LPS-induced inflammatory stimulus: evidence in Parkinson’s disease patients. Neurobiol Dis 41(1):177–188. https://doi.org/10.1016/j.nbd.2010.09.005
Martire S, Mosca L, d'Erme M (2015) PARP-1 involvement in neurodegeneration: a focus on Alzheimer’s and Parkinson’s diseases. Mech Ageing Dev 146-148:53–64. https://doi.org/10.1016/j.mad.2015.04.001
Song L, Pei L, Yao S, Wu Y, Shang Y (2017) NLRP3 inflammasome in neurological diseases, from functions to therapies. Front Cell Neurosci 11:63. https://doi.org/10.3389/fncel.2017.00063
Zhang YW, Thompson R, Zhang H, Xu H (2011) APP processing in Alzheimer’s disease. Mol Brain 4:3. https://doi.org/10.1186/1756-6606-4-3
Morris G, Berk M, Walder K, Maes M (2015) Central pathways causing fatigue in neuro-inflammatory and autoimmune illnesses. BMC Med 13(1):1–23. https://doi.org/10.1186/s12916-014-0259-2
Rodríguez-Vargas JM, Ruiz-Magaña MJ, Ruiz-Ruiz C, Majuelos-Melguizo J, Peralta-Leal A, Rodríguez MI, Muñoz-Gámez JA, de Almodóvar MR et al (2012) ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res 22(7):1181–1198. https://doi.org/10.1038/cr.2012.70
Abais JM, Xia M, Zhang Y, Boini KM, Li P-L (2015) Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal 22(13):1111–1129. https://doi.org/10.1089/ars.2014.5994
Cao JY, Dixon SJ (2016) Mechanisms of ferroptosis. Cell Mol Life Sci 73(11):2195–2209. https://doi.org/10.1007/s00018-016-2194-1
Martínez-Zamudio RI, Ha HC (2014) PARP1 enhances inflammatory cytokine expression by alteration of promoter chromatin structure in microglia. Brain Behav 4(4):552–565. https://doi.org/10.1002/brb3.239
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D (2016) Ferroptosis: process and function. Cell Death Differ 23(3):369–379. https://doi.org/10.1038/cdd.2015.158
Szebeni A, Szebeni K, DiPeri TP, Johnson LA, Stockmeier CA, Crawford JD, Chandley MJ, Hernandez LJ et al (2017) Elevated DNA oxidation and DNA repair enzyme expression in brain white matter in major depressive disorder. Int J Neuropsychopharmacol 20(5):363–373. https://doi.org/10.1093/ijnp/pyw114
Kaufmann FN, Costa AP, Ghisleni G, Diaz AP, Rodrigues ALS, Peluffo H, Kaster MP (2017) NLRP3 inflammasome-driven pathways in depression: clinical and preclinical findings. Brain Behav Immun 64:367–383. https://doi.org/10.1016/j.bbi.2017.03.002
Kim YK, Na KS, Myint AM, Leonard BE (2016) The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Prog Neuro-Psychopharmacol Biol Psychiatry 64:277–284. https://doi.org/10.1016/j.pnpbp.2015.06.008
Zhao Z, Xu J, Chen J, Kim S, Reimers M, Bacanu S-A, Yu H, Liu C et al (2015) Transcriptome sequencing and genome-wide association analyses reveal lysosomal function and actin cytoskeleton remodeling in schizophrenia and bipolar disorder. Mol Psychiatry 20(5):563–572. https://doi.org/10.1038/mp.2014.82
Borsini A, Zunszain PA, Thuret S, Pariante CM (2015) The role of inflammatory cytokines as key modulators of neurogenesis. Trends Neurosci 38(3):145–157. https://doi.org/10.1016/j.tins.2014.12.006
Simonaro CM (2016) Lysosomes, lysosomal storage diseases, and inflammation. J Inborn Errors Metab Screen 4:2326409816650465. https://doi.org/10.1177/2326409816650465
Maes M, Van de Vyvere J, Vandoolaeghe E, Bril T, Demedts P, Wauters A, Neels H (1996) Alterations in iron metabolism and the erythron in major depression: further evidence for a chronic inflammatory process. J Affect Disord 40(1):23–33. https://doi.org/10.1016/0165-0327(96)00038-9
Serata D, Del Casale A, Rapinesi C, Mancinelli I, Pompili P, Kotzalidis GD, Aimati L, Savoja V et al (2012) Hemochromatosis-induced bipolar disorder: a case report. Gen Hosp Psychiatry 34(1):101e101–101e103. https://doi.org/10.1016/j.genhosppsych.2011.04.013
Cutler P (1994) Iron overload and psychiatric illness. Can J Psychiatr 39(1):8–11
Feifel D, Young CW (1997) Iron overload among a psychiatric outpatient population. J Clin Psychiatry 58(2):74–78
Casanova MF, Comparini SO, Kim RW, Kleinman JE (1992) Staining intensity of brain iron in patients with schizophrenia: a postmortem study. J Neuropsychiatry Clin Neurosci 4(1):36–41. https://doi.org/10.1176/jnp.4.1.36
Urrutia P, Mena N, Nunez M (2014) The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front Pharmacol 5:38. https://doi.org/10.3389/fphar.2014.00038
Silva B, Faustino P (2015) An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim Biophys Acta 1852(7):1347–1359. https://doi.org/10.1016/j.bbadis.2015.03.011
Bresgen N, Jaksch H, Lacher H, Ohlenschlager I, Uchida K, Eckl PM (2010) Iron-mediated oxidative stress plays an essential role in ferritin-induced cell death. Free Radic Biol Med 48(10):1347–1357. https://doi.org/10.1016/j.freeradbiomed.2010.02.019
Chase A (2015) Neuroinflammation: targeting neuroinflammation through inhibition of NLRP3. Nat Rev Neurol 11(4):186–186. https://doi.org/10.1038/nrneurol.2015.31
Orozco S, Oberst A (2017) RIPK3 in cell death and inflammation: the good, the bad, and the ugly. Immunol Rev 277(1):102–112. https://doi.org/10.1111/imr.12536
Moriwaki K, Chan FK (2017) The inflammatory signal adaptor RIPK3: functions beyond necroptosis. Int Rev Cell Mol Biol 328:253–275. https://doi.org/10.1016/bs.ircmb.2016.08.007
Fuster-Matanzo A, Llorens-Martin M, Hernandez F, Avila J (2013) Role of neuroinflammation in adult neurogenesis and Alzheimer disease: therapeutic approaches. Mediat Inflamm 2013:260925. https://doi.org/10.1155/2013/260925
Miller AH, Raison CL (2016) The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 16(1):22–34. https://doi.org/10.1038/nri.2015.5
Walker NI, Harmon BV, Gobe GC, Kerr JF (1988) Patterns of cell death. Methods Achiev Exp Pathol 13:18–54
Nicotera P, Leist M, Ferrando-May E (1998) Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol Lett 102–103:139–142
Han J, Zhong C-Q, Zhang D-W (2011) Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat Immunol 12(12):1143–1149
Günther C, Neumann H, Neurath MF, Becker C (2013) Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut 62(7):1062–1071. https://doi.org/10.1136/gutjnl-2011-301364
Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P (1997) Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med 185(8):1481–1486
Liu T, Bao YH, Wang Y, Jiang JY (2015) The role of necroptosis in neurosurgical diseases. Braz J Med Biol Res 48(4):292–298. https://doi.org/10.1590/1414-431X20144310
Liu X, Shi F, Li Y, Yu X, Peng S, Li W, Luo X, Cao Y (2016) Post-translational modifications as key regulators of TNF-induced necroptosis. Cell Death Dis 7(7):e2293. https://doi.org/10.1038/cddis.2016.197
Zhang L, Blackwell K, Workman LM, Chen S, Pope MR, Janz S, Habelhah H (2015) RIP1 cleavage in the kinase domain regulates TRAIL-induced NF-kappaB activation and lymphoma survival. Mol Cell Biol 35(19):3324–3338. https://doi.org/10.1128/mcb.00692-15
Blackwell K, Zhang L, Workman LM, Ting AT, Iwai K, Habelhah H (2013) Two coordinated mechanisms underlie tumor necrosis factor alpha-induced immediate and delayed IkappaB kinase activation. Mol Cell Biol 33(10):1901–1915. https://doi.org/10.1128/mcb.01416-12
Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ (2006) Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 22(2):245–257. https://doi.org/10.1016/j.molcel.2006.03.026
Dondelinger Y, Darding M, Bertrand MJ, Walczak H (2016) Poly-ubiquitination in TNFR1-mediated necroptosis. Cell Mol Life Sci: CMLS 73(11–12):2165–2176. https://doi.org/10.1007/s00018-016-2191-4
Ikeda F (2015) Linear ubiquitination signals in adaptive immune responses. Immunol Rev 266(1):222–236. https://doi.org/10.1111/imr.12300
Humphries F, Yang S, Wang B, Moynagh PN (2015) RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ 22(2):225–236. https://doi.org/10.1038/cdd.2014.126
Lin Y, Devin A, Rodriguez Y, Liu ZG (1999) Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 13(19):2514–2526
Moriwaki K, Chan FK-M (2013) RIP3: a molecular switch for necrosis and inflammation. Genes Dev 27(15):1640–1649. https://doi.org/10.1101/gad.223321.113
Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG (1993) Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res 53(17):3976–3985
Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS et al (1995) Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 81(5):801–809
Margolin N, Raybuck SA, Wilson KP, Chen W, Fox T, Gu Y, Livingston DJ (1997) Substrate and inhibitor specificity of interleukin-1 beta-converting enzyme and related caspases. J Biol Chem 272(11):7223–7228
Soldani C, Lazze MC, Bottone MG, Tognon G, Biggiogera M, Pellicciari CE, Scovassi AI (2001) Poly(ADP-ribose) polymerase cleavage during apoptosis: when and where? Exp Cell Res 269(2):193–201. https://doi.org/10.1006/excr.2001.5293
Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC (1994) Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371(6495):346–347. https://doi.org/10.1038/371346a0
D’Amours D, Sallmann FR, Dixit VM, Poirier GG (2001) Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: implications for apoptosis. J Cell Sci 114(Pt 20):3771–3778
Hayakawa R, Hayakawa T, Takeda K, Ichijo H (2012) Therapeutic targets in the ASK1-dependent stress signaling pathways. Proc Jpn Acad Ser B, Phys Biol Sci 88(8):434–453. https://doi.org/10.2183/pjab.88.434
Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K et al (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17(9):2596–2606. https://doi.org/10.1093/emboj/17.9.2596
Katagiri K, Matsuzawa A, Ichijo H (2010) Regulation of apoptosis signal-regulating kinase 1 in redox signaling. Methods Enzymol 474:277–288. https://doi.org/10.1016/s0076-6879(10)74016-7
Fujino G, Noguchi T, Takeda K, Ichijo H (2006) Thioredoxin and protein kinases in redox signaling. Semin Cancer Biol 16(6):427–435. https://doi.org/10.1016/j.semcancer.2006.09.003
Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, Choi AM (2007) Mechanisms of cell death in oxidative stress. Antioxid Redox Signal 9(1):49–89. https://doi.org/10.1089/ars.2007.9.49
Shen HM, Lin Y, Choksi S, Tran J, Jin T, Chang L, Karin M, Zhang J et al (2004) Essential roles of receptor-interacting protein and TRAF2 in oxidative stress-induced cell death. Mol Cell Biol 24(13):5914–5922. https://doi.org/10.1128/MCB.24.13.5914-5922.2004
Morgan MJ, Kim YS, Liu Z (2007) Lipid rafts and oxidative stress-induced cell death. Antioxid Redox Signal 9(9):1471–1483. https://doi.org/10.1089/ars.2007.1658
Wu YT, Zhang S, Kim YS, Tan HL, Whiteman M, Ong CN, Liu ZG, Ichijo H et al (2008) Signaling pathways from membrane lipid rafts to JNK1 activation in reactive nitrogen species-induced non-apoptotic cell death. Cell Death Differ 15(2):386–397. https://doi.org/10.1038/sj.cdd.4402273
Liu F, Jiang N, Xiao ZY, Cheng JP, Mei YZ, Zheng P, Wang L, Zhang XR et al (2016) Effects of poly (ADP-ribose) polymerase-1 (PARP-1) inhibition on sulfur mustard-induced cutaneous injuries in vitro and in vivo. PeerJ 4:e1890. https://doi.org/10.7717/peerj.1890
Bidovec K, Bozic J, Dolenc I, Turk B, Turk V, Stoka V (2017) Tumor necrosis factor-alpha induced apoptosis in U937 cells promotes cathepsin D-independent stefin B degradation. J Cell Biochem. https://doi.org/10.1002/jcb.26152
Guicciardi ME, Deussing J, Miyoshi H, Bronk SF, Svingen PA, Peters C, Kaufmann SH, Gores GJ (2000) Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest 106(9):1127–1137. https://doi.org/10.1172/JCI9914
Deiss LP, Galinka H, Berissi H, Cohen O, Kimchi A (1996) Cathepsin D protease mediates programmed cell death induced by interferon-gamma, Fas/APO-1 and TNF-alpha. EMBO J 15(15):3861–3870
Aits S, Jaattela M (2013) Lysosomal cell death at a glance. J Cell Sci 126(Pt 9):1905–1912. https://doi.org/10.1242/jcs.091181
Castino R, Bellio N, Nicotra G, Follo C, Trincheri NF, Isidoro C (2007) Cathepsin D-Bax death pathway in oxidative stressed neuroblastoma cells. Free Radic Biol Med 42(9):1305–1316. https://doi.org/10.1016/j.freeradbiomed.2006.12.030
Cirman T, Oresic K, Mazovec GD, Turk V, Reed JC, Myers RM, Salvesen GS, Turk B (2004) Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J Biol Chem 279(5):3578–3587. https://doi.org/10.1074/jbc.M308347200
Heinrich M, Neumeyer J, Jakob M, Hallas C, Tchikov V, Winoto-Morbach S, Wickel M, Schneider-Brachert W et al (2004) Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ 11(5):550–563. https://doi.org/10.1038/sj.cdd.4401382
Fiandalo MV, Schwarze SR, Kyprianou N (2013) Proteasomal regulation of caspase-8 in cancer cell apoptosis. Apoptosis: Int J Program Cell death 18(6):766–776. https://doi.org/10.1007/s10495-013-0821-y
O’Reilly E, Tirincsi A, Logue SE, Szegezdi E (2016) The Janus face of death receptor signaling during tumor immunoediting. Front Immunol 7:446. https://doi.org/10.3389/fimmu.2016.00446
Kim M, Hernandez L, Annunziata CM (2016) Caspase 8 expression may determine the survival of women with ovarian cancer. Cell Death Dis 7:e2045. https://doi.org/10.1038/cddis.2015.398
Moquin DM, McQuade T, Chan FK (2013) CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One 8(10):e76841. https://doi.org/10.1371/journal.pone.0076841
Vanden Berghe T, Kaiser WJ, Bertrand MJ, Vandenabeele P (2015) Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol Cell Oncol 2(4):e975093. https://doi.org/10.4161/23723556.2014.975093
Wang XD, Li CY, Jiang MM, Li D, Wen P, Song X, Chen JD, Guo LX et al (2016) Induction of apoptosis in human leukemia cells through an intrinsic pathway by cathachunine, a unique alkaloid isolated from Catharanthus roseus. Phytomedicine 23(6):641–653. https://doi.org/10.1016/j.phymed.2016.03.003
Redza-Dutordoir M, Averill-Bates DA (2016) Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta 1863(12):2977–2992. https://doi.org/10.1016/j.bbamcr.2016.09.012
Schenk B, Fulda S (2015) Reactive oxygen species regulate Smac mimetic/TNFalpha-induced necroptotic signaling and cell death. Oncogene 34(47):5796–5806. https://doi.org/10.1038/onc.2015.35
Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, Cai Q, Yang ZH et al (2017) RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun 8:14329. https://doi.org/10.1038/ncomms14329
Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T (2011) TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis 2:e230. https://doi.org/10.1038/cddis.2011.111
Vanlangenakker N, Vanden Berghe T, Vandenabeele P (2012) Many stimuli pull the necrotic trigger, an overview. Cell Death Differ 19(1):75–86. https://doi.org/10.1038/cdd.2011.164
Lemaire C, Andreau K, Souvannavong V, Adam A (1998) Inhibition of caspase activity induces a switch from apoptosis to necrosis. FEBS Lett 425(2):266–270
Aikin R, Rosenberg L, Paraskevas S, Maysinger D (2004) Inhibition of caspase-mediated PARP-1 cleavage results in increased necrosis in isolated islets of Langerhans. J Mol Med (Berl) 82(6):389–397. https://doi.org/10.1007/s00109-004-0540-5
Eguchi Y, Shimizu S, Tsujimoto Y (1997) Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res 57(10):1835–1840
Festjens N, Vanden Berghe T, Vandenabeele P (2006) Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta 1757(9–10):1371–1387. https://doi.org/10.1016/j.bbabio.2006.06.014
Yu C, Kim BS, Kim E (2016) FAF1 mediates regulated necrosis through PARP1 activation upon oxidative stress leading to dopaminergic neurodegeneration. Cell Death Differ 23(11):1873–1885. https://doi.org/10.1038/cdd.2016.99
Baritaud M, Cabon L, Delavallee L, Galan-Malo P, Gilles ME, Brunelle-Navas MN, Susin SA (2012) AIF-mediated caspase-independent necroptosis requires ATM and DNA-PK-induced histone H2AX Ser139 phosphorylation. Cell Death Dis 3:e390. https://doi.org/10.1038/cddis.2012.120
Xu Y, Huang S, Liu Z-G, Han J (2006) Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. J Biol Chem 281(13):8788–8795. https://doi.org/10.1074/jbc.M508135200
Douglas DL, Baines CP (2014) PARP1-mediated necrosis is dependent on parallel JNK and Ca(2+)/calpain pathways. J Cell Sci 127(19):4134–4145. https://doi.org/10.1242/jcs.128009
Harijith A, Ebenezer DL, Natarajan V (2014) Reactive oxygen species at the crossroads of inflammasome and inflammation. Front Physiol 5:352. https://doi.org/10.3389/fphys.2014.00352
Vanaja SK, Rathinam VA, Fitzgerald KA (2015) Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol 25(5):308–315. https://doi.org/10.1016/j.tcb.2014.12.009
Dorfleutner A, Chu L, Stehlik C (2015) Inhibiting the inflammasome: one domain at a time. Immunol Rev 265(1):205–216. https://doi.org/10.1111/imr.12290
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B et al (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526(7575):666–671. https://doi.org/10.1038/nature15541
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T et al (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526(7575):660–665. https://doi.org/10.1038/nature15514
Fink SL, Bergsbaken T, Cookson BT (2008) Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci U S A 105(11):4312–4317. https://doi.org/10.1073/pnas.0707370105
Ramesh G, MacLean AG, Philipp MT (2013) Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediat Inflamm 2013:20. https://doi.org/10.1155/2013/480739
Koedel U, Frankenberg T, Kirschnek S, Obermaier B, Hacker H, Paul R, Hacker G (2009) Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental pneumococcal meningitis. PLoS Pathog 5(5):e1000461. https://doi.org/10.1371/journal.ppat.1000461
Yang J, Liu Z, Xiao TS (2017) Post-translational regulation of inflammasomes. Cell Mol Immunol 14(1):65–79. https://doi.org/10.1038/cmi.2016.29
Yang CA, Chiang BL (2015) Inflammasomes and human autoimmunity: a comprehensive review. J Autoimmun 61:1–8. https://doi.org/10.1016/j.jaut.2015.05.001
Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT (2014) Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci 8:315. https://doi.org/10.3389/fnins.2014.00315
Haasken S, Sutterwala FS (2013) Damage control: management of cellular stress by the NLRP3 inflammasome. Eur J Immunol 43(8):2003–2005. https://doi.org/10.1002/eji.201343848
Hampton MB, Fadeel B, Orrenius S (1998) Redox regulation of the caspases during apoptosisa. Ann N Y Acad Sci 854(1):328–335. https://doi.org/10.1111/j.1749-6632.1998.tb09913.x
Reinheckel T (2013) On the road to inflammation: linking lysosome disruption, lysosomal protease release and necrotic death of immune cells. Cell Cycle (Georgetown, Tex) 12(13):1994. https://doi.org/10.4161/cc.25316
Lima H Jr, Jacobson LS, Goldberg MF, Chandran K, Diaz-Griffero F, Lisanti MP, Brojatsch J (2013) Role of lysosome rupture in controlling Nlrp3 signaling and necrotic cell death. Cell Cycle (Georgetown, Tex) 12(12):1868–1878. https://doi.org/10.4161/cc.24903
Guicciardi ME, Gores GJ (2013) Complete lysosomal disruption: a route to necrosis, not to the inflammasome. Cell Cycle (Georgetown, Tex) 12(13):1995. https://doi.org/10.4161/cc.25317
Guicciardi ME, Leist M, Gores GJ (2004) Lysosomes in cell death. Oncogene 23(16):2881–2890. https://doi.org/10.1038/sj.onc.1207512
Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Hoyer-Hansen M, Weber E, Multhoff G et al (2004) Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med 200(4):425–435. https://doi.org/10.1084/jem.20040531
Zhao M, Antunes F, Eaton JW, Brunk UT (2003) Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis. Eur J Biochem / FEBS 270(18):3778–3786
Barbouti A, Amorgianiotis C, Kolettas E, Kanavaros P, Galaris D (2007) Hydrogen peroxide inhibits caspase-dependent apoptosis by inactivating procaspase-9 in an iron-dependent manner. Free Radic Biol Med 43(10):1377–1387. https://doi.org/10.1016/j.freeradbiomed.2007.06.020
Brunk UT, Eaton JW (2007) Peroxide hormesis? A commentary on “Hydrogen peroxide inhibits caspase-dependent apoptosis by inactivating procaspase-9 in an iron-dependent manner”. Free Radic Biol Med 43(10):1372–1373. https://doi.org/10.1016/j.freeradbiomed.2007.08.008
Kool M, Soullie T, van Nimwegen M, Willart MA, Muskens F, Jung S, Hoogsteden HC, Hammad H et al (2008) Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J Exp Med 205(4):869–882. https://doi.org/10.1084/jem.20071087
Kool M, Petrilli V, De Smedt T, Rolaz A, Hammad H, van Nimwegen M, Bergen IM, Castillo R et al (2008) Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol (Baltimore, Md : 1950) 181(6):3755–3759
Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9(8):847–856. https://doi.org/10.1038/ni.1631
Lin Y, Epstein DL, Liton PB (2010) Intralysosomal iron induces lysosomal membrane permeabilization and cathepsin D-mediated cell death in trabecular meshwork cells exposed to oxidative stress. Invest Ophthalmol Vis Sci 51(12):6483–6495. https://doi.org/10.1167/iovs.10-5410
Kurz T, Terman A, Gustafsson B, Brunk UT (2008) Lysosomes in iron metabolism, ageing and apoptosis. Histochem Cell Biol 129(4):389–406. https://doi.org/10.1007/s00418-008-0394-y
Kurz T, Gustafsson B, Brunk UT (2006) Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. FEBS J 273(13):3106–3117. https://doi.org/10.1111/j.1742-4658.2006.05321.x
Yu Z, Persson HL, Eaton JW, Brunk UT (2003) Intralysosomal iron: a major determinant of oxidant-induced cell death. Free Radic Biol Med 34(10):1243–1252
Bresgen N, Eckl PM (2015) Oxidative stress and the homeodynamics of iron metabolism. Biomol Ther 5(2):808–847. https://doi.org/10.3390/biom5020808
Galaris D, Pantopoulos K (2008) Oxidative stress and iron homeostasis: mechanistic and health aspects. Crit Rev Clin Lab Sci 45(1):1–23. https://doi.org/10.1080/10408360701713104
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149(5):1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA et al (2014) Regulation of ferroptotic cancer cell death by GPX4. Cell 156(1–2):317–331. https://doi.org/10.1016/j.cell.2013.12.010
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR (2016) Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113(34):E4966–E4975. https://doi.org/10.1073/pnas.1603244113
Yang WS, Stockwell BR (2016) Ferroptosis: death by lipid peroxidation. Trends Cell Biol 26(3):165–176. https://doi.org/10.1016/j.tcb.2015.10.014
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X (2015) Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 59(2):298–308. https://doi.org/10.1016/j.molcel.2015.06.011
Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, Suzuki T, Mori M et al (2016) An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J 473(6):769–777. https://doi.org/10.1042/bj20150658
Guo J, Chen Q, Tang J, Zhang J, Tao Y, Li L, Zhu G, Feng H et al (2015) Minocycline-induced attenuation of iron overload and brain injury after experimental germinal matrix hemorrhage. Brain Res 1594:115–124. https://doi.org/10.1016/j.brainres.2014.10.046
Krainz T, Gaschler MM, Lim C, Sacher JR, Stockwell BR, Wipf P (2016) A mitochondrial-targeted nitroxide is a potent inhibitor of ferroptosis. ACS Central Sci 2(9):653–659. https://doi.org/10.1021/acscentsci.6b00199
Neitemeier S, Jelinek A, Laino V, Hoffmann L, Eisenbach I, Eying R, Ganjam GK, Dolga AM et al (2017) BID links ferroptosis to mitochondrial cell death pathways. Redox Biol 12:558–570. https://doi.org/10.1016/j.redox.2017.03.007
D'Herde K, Krysko DV (2017) Ferroptosis: oxidized PEs trigger death. Nat Chem Biol 13(1):4–5. https://doi.org/10.1038/nchembio.2261
Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, Dar HH, Liu B et al (2017) Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13(1):81–90. https://doi.org/10.1038/nchembio.2238
Latunde-Dada GO (2017) Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta 1861(8):1893–1900. https://doi.org/10.1016/j.bbagen.2017.05.019
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J et al (2017) ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13(1):91–98. https://doi.org/10.1038/nchembio.2239
Doll S, Conrad M (2017) Iron and ferroptosis: a still ill-defined liaison. IUBMB Life 69(6):423–434. https://doi.org/10.1002/iub.1616
Muller T, Dewitz C, Schmitz J, Schroder AS, Brasen JH, Stockwell BR, Murphy JM, Kunzendorf U et al (2017) Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci: CMLS. https://doi.org/10.1007/s00018-017-2547-4
Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol 8(4):382–397. https://doi.org/10.1016/s1474-4422(09)70062-6
Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T (1994) Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett 165(1–2):208–210
McGeer EG, McGeer PL (2003) Inflammatory processes in Alzheimer’s disease. Prog Neuro-Psychopharmacol Biol Psychiatry 27(5):741–749. https://doi.org/10.1016/S0278-5846(03)00124-6
Zhao M, Cribbs DH, Anderson AJ, Cummings BJ, Su JH, Wasserman AJ, Cotman CW (2003) The induction of the TNFalpha death domain signaling pathway in Alzheimer’s disease brain. Neurochem Res 28(2):307–318
Cheng X, Yang L, He P, Li R, Shen Y (2010) Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer’s disease and non-demented patients. J Alzheimer's Dis: JAD 19(2):621–630. https://doi.org/10.3233/jad-2010-1253
Dong Y, Dekens WD, De Deyn PP, Naudé JP, Eisel LU (2015) Targeting of tumor necrosis factor alpha receptors as a therapeutic strategy for neurodegenerative disorders. Antibodies 4(4):369–408. https://doi.org/10.3390/antib4040369
Diniz BS, Teixeira AL, Ojopi EB, Talib LL, Mendonca VA, Gattaz WF, Forlenza OV (2010) Higher serum sTNFR1 level predicts conversion from mild cognitive impairment to Alzheimer’s disease. J Alzheimer's Dis: JAD 22(4):1305–1311. https://doi.org/10.3233/jad-2010-100921
Rocha NP, Teixeira AL, Scalzo PL, Barbosa IG, de Sousa MS, Morato IB, Vieira EL, Christo PP et al (2014) Plasma levels of soluble tumor necrosis factor receptors are associated with cognitive performance in Parkinson’s disease. Mov Disord 29(4):527–531. https://doi.org/10.1002/mds.25752
Himmerich H, Fulda S, Linseisen J, Seiler H, Wolfram G, Himmerich S, Gedrich K, Kloiber S et al (2008) Depression, comorbidities and the TNF-alpha system. Eur Psychiatry 23(6):421–429. https://doi.org/10.1016/j.eurpsy.2008.03.013
Maes M, Berk M, Goehler L, Song C, Anderson G, Galecki P (2012) Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med 10:66
Soczynska JK, Kennedy SH, Goldstein BI, Lachowski A, Woldeyohannes HO, McIntyre RS (2009) The effect of tumor necrosis factor antagonists on mood and mental health-associated quality of life: novel hypothesis-driven treatments for bipolar depression? Neurotoxicology 30(4):497–521. https://doi.org/10.1016/j.neuro.2009.03.004
Hoseth EZ, Ueland T, Dieset I, Birnbaum R, Shin JH, Kleinman JE, Hyde TM, Morch RH et al (2017) A study of TNF pathway activation in schizophrenia and bipolar disorder in plasma and brain tissue. Schizophr Bull. https://doi.org/10.1093/schbul/sbw183
O’Brien SM, Scully P, Dinan TG (2008) Increased tumor necrosis factor-alpha concentrations with interleukin-4 concentrations in exacerbations of schizophrenia. Psychiatry Res 160(3):256–262. https://doi.org/10.1016/j.psychres.2007.11.014
Peng J, Andersen JK (2003) The role of c-Jun N-terminal kinase (JNK) in Parkinson’s disease. IUBMB Life 55(4–5):267–271. https://doi.org/10.1080/1521654031000121666
Yarza R, Vela S, Solas M, Ramirez MJ (2015) c-Jun N-terminal kinase (JNK) signaling as a therapeutic target for alzheimer’s disease. Front Pharmacol 6:321. https://doi.org/10.3389/fphar.2015.00321
Akhter R, Sanphui P, Das H, Saha P, Biswas SC (2015) The regulation of p53 up-regulated modulator of apoptosis by JNK/c-Jun pathway in beta-amyloid-induced neuron death. J Neurochem 134(6):1091–1103. https://doi.org/10.1111/jnc.13128
Coffey ET (2014) Nuclear and cytosolic JNK signalling in neurons. Nat Rev Neurosci 15(5):285–299. https://doi.org/10.1038/nrn3729
Bjorkblom B, Padzik A, Mohammad H, Westerlund N, Komulainen E, Hollos P, Parviainen L, Papageorgiou AC et al (2012) c-Jun N-terminal kinase phosphorylation of MARCKSL1 determines actin stability and migration in neurons and in cancer cells. Mol Cell Biol 32(17):3513–3526. https://doi.org/10.1128/mcb.00713-12
Podkowa M, Zhao X, Chow CW, Coffey ET, Davis RJ, Attisano L (2010) Microtubule stabilization by bone morphogenetic protein receptor-mediated scaffolding of c-Jun N-terminal kinase promotes dendrite formation. Mol Cell Biol 30(9):2241–2250. https://doi.org/10.1128/mcb.01166-09
Komulainen E, Zdrojewska J, Freemantle E, Mohammad H, Kulesskaya N, Deshpande P, Marchisella F, Mysore R et al (2014) JNK1 controls dendritic field size in L2/3 and L5 of the motor cortex, constrains soma size, and influences fine motor coordination. Front Cell Neurosci 8:272. https://doi.org/10.3389/fncel.2014.00272
Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H et al (2008) Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 358(7):667–675. https://doi.org/10.1056/NEJMoa075974
Winchester CL, Ohzeki H, Vouyiouklis DA, Thompson R, Penninger JM, Yamagami K, Norrie JD, Hunter R et al (2012) Converging evidence that sequence variations in the novel candidate gene MAP2K7 (MKK7) are functionally associated with schizophrenia. Hum Mol Genet 21(22):4910–4921. https://doi.org/10.1093/hmg/dds331
McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE et al (2009) Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet 41(11):1223–1227. https://doi.org/10.1038/ng.474
Pavlowsky A, Gianfelice A, Pallotto M, Zanchi A, Vara H, Khelfaoui M, Valnegri P, Rezai X et al (2010) A postsynaptic signaling pathway that may account for the cognitive defect due to IL1RAPL1 mutation. Curr Biol: CB 20(2):103–115. https://doi.org/10.1016/j.cub.2009.12.030
Kunde SA, Rademacher N, Tzschach A, Wiedersberg E, Ullmann R, Kalscheuer VM, Shoichet SA (2013) Characterisation of de novo MAPK10/JNK3 truncation mutations associated with cognitive disorders in two unrelated patients. Hum Genet 132(4):461–471. https://doi.org/10.1007/s00439-012-1260-5
Shiizaki S, Naguro I, Ichijo H (2013) Activation mechanisms of ASK1 in response to various stresses and its significance in intracellular signaling. Adv Biol Regul 53(1):135–144. https://doi.org/10.1016/j.jbior.2012.09.006
Soga M, Matsuzawa A, Ichijo H (2012) Oxidative stress-induced diseases via the ASK1 signaling pathway. Int J Cell Biol 2012:439587. https://doi.org/10.1155/2012/439587
Harada C, Nakamura K, Namekata K, Okumura A, Mitamura Y, Iizuka Y, Kashiwagi K, Yoshida K et al (2006) Role of apoptosis signal-regulating kinase 1 in stress-induced neural cell apoptosis in vivo. Am J Pathol 168(1):261–269. https://doi.org/10.2353/ajpath.2006.050765
Eaton GJ, Zhang QS, Diallo C, Matsuzawa A, Ichijo H, Steinbeck MJ, Freeman TA (2014) Inhibition of apoptosis signal-regulating kinase 1 enhances endochondral bone formation by increasing chondrocyte survival. Cell Death Dis 5:e1522. https://doi.org/10.1038/cddis.2014.480
Kadowaki H, Nishitoh H, Urano F, Sadamitsu C, Matsuzawa A, Takeda K, Masutani H, Yodoi J et al (2005) Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ 12(1):19–24. https://doi.org/10.1038/sj.cdd.4401528
Hu X, Weng Z, Chu CT, Zhang L, Cao G, Gao Y, Signore A, Zhu J et al (2011) Peroxiredoxin-2 protects against 6-hydroxydopamine-induced dopaminergic neurodegeneration via attenuation of the apoptosis signal-regulating kinase (ASK1) signaling cascade. J Neurosci 31(1):247–261. https://doi.org/10.1523/jneurosci.4589-10.2011
Guo X, Harada C, Namekata K, Matsuzawa A, Camps M, Ji H, Swinnen D, Jorand-Lebrun C et al (2010) Regulation of the severity of neuroinflammation and demyelination by TLR-ASK1-p38 pathway. EMBO Mol Med 2(12):504–515. https://doi.org/10.1002/emmm.201000103
Chaitanya GV, Alexander JS, Babu PP (2010) PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal: CCS 8:31–31. https://doi.org/10.1186/1478-811X-8-31
Krishnakumar R, Kraus WL (2010) The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell 39(1):8–24. https://doi.org/10.1016/j.molcel.2010.06.017
Kraus WL, Hottiger MO (2013) PARP-1 and gene regulation: progress and puzzles. Mol Asp Med 34(6):1109–1123. https://doi.org/10.1016/j.mam.2013.01.005
Rosado MM, Bennici E, Novelli F, Pioli C (2013) Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology 139(4):428–437. https://doi.org/10.1111/imm.12099
Phulwani NK, Esen N, Syed MM, Kielian T (2008) TLR2 expression in astrocytes is induced by TNF-alpha- and NF-kappa B-dependent pathways. J Immunol (Baltimore, Md : 1950) 181(6):3841–3849
Chiarugi A, Moskowitz MA (2003) Poly(ADP-ribose) polymerase-1 activity promotes NF-kappaB-driven transcription and microglial activation: implication for neurodegenerative disorders. J Neurochem 85(2):306–317
Phulwani NK, Kielian T (2008) Poly (ADP-ribose) polymerases (PARPs) 1-3 regulate astrocyte activation. J Neurochem 106(2):578–590. https://doi.org/10.1111/j.1471-4159.2008.05403.x
Nakajima H, Nagaso H, Kakui N, Ishikawa M, Hiranuma T, Hoshiko S (2004) Critical role of the automodification of poly(ADP-ribose) polymerase-1 in nuclear factor-kappaB-dependent gene expression in primary cultured mouse glial cells. J Biol Chem 279(41):42774–42786. https://doi.org/10.1074/jbc.M407923200
Tang KS, Suh SW, Alano CC, Shao Z, Hunt WT, Swanson RA, Anderson CM (2010) Astrocytic poly(ADP-ribose) polymerase-1 activation leads to bioenergetic depletion and inhibition of glutamate uptake capacity. Glia 58(4):446–457. https://doi.org/10.1002/glia.20936
Visochek L, Steingart RA, Vulih-Shultzman I, Klein R, Priel E, Gozes I, Cohen-Armon M (2005) PolyADP-ribosylation is involved in neurotrophic activity. J Neurosci 25(32):7420–7428. https://doi.org/10.1523/jneurosci.0333-05.2005
Homburg S, Visochek L, Moran N, Dantzer F, Priel E, Asculai E, Schwartz D, Rotter V et al (2000) A fast signal-induced activation of Poly(ADP-ribose) polymerase: a novel downstream target of phospholipase c. J Cell Biol 150(2):293–307
Inaba H, Tsukagoshi A, Kida S (2015) PARP-1 activity is required for the reconsolidation and extinction of contextual fear memory. Mol Brain 8(1):63. https://doi.org/10.1186/s13041-015-0153-7
Alfonso-Loeches S, Urena-Peralta JR, Morillo-Bargues MJ, Oliver-De La Cruz J, Guerri C (2014) Role of mitochondria ROS generation in ethanol-induced NLRP3 inflammasome activation and cell death in astroglial cells. Front Cell Neurosci 8:216. https://doi.org/10.3389/fncel.2014.00216
Kim JY, Paton JC, Briles DE, Rhee DK, Pyo S (2015) Streptococcus pneumoniae induces pyroptosis through the regulation of autophagy in murine microglia. Oncotarget 6(42):44161–44178. 10.18632/oncotarget.6592
Fann DY, Lee SY, Manzanero S, Tang SC, Gelderblom M, Chunduri P, Bernreuther C, Glatzel M et al (2013) Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis 4:e790. https://doi.org/10.1038/cddis.2013.326
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D et al (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493(7434):674–678. https://doi.org/10.1038/nature11729
Johann S, Heitzer M, Kanagaratnam M, Goswami A, Rizo T, Weis J, Troost D, Beyer C (2015) NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 63(12):2260–2273. https://doi.org/10.1002/glia.22891
Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, Rainone V, Nemni R et al (2016) The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener 11:23. https://doi.org/10.1186/s13024-016-0088-1
Huang WX, Huang P, Hillert J (2004) Increased expression of caspase-1 and interleukin-18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult Scler 10(5):482–487. https://doi.org/10.1191/1352458504ms1071oa
Inoue M, Williams KL, Gunn MD, Shinohara ML (2012) NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 109(26):10480–10485. https://doi.org/10.1073/pnas.1201836109
Debye B, Schmulling L, Zhou L, Rune G, Beyer C, Johann S (2016) Neurodegeneration and NLRP3 inflammasome expression in the anterior thalamus of SOD1(G93A) ALS mice. Brain Pathol. https://doi.org/10.1111/bpa.12467
de Rivero Vaccari JP, Brand F 3rd, Adamczak S, Lee SW, Perez-Barcena J, Wang MY, Bullock MR, Dietrich WD et al (2016) Exosome-mediated inflammasome signaling after central nervous system injury. J Neurochem 136(Suppl 1):39–48. https://doi.org/10.1111/jnc.13036
Dinarello CA, Simon A, van der Meer JW (2012) Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 11(8):633–652. https://doi.org/10.1038/nrd3800
Arend WP, Palmer G, Gabay C (2008) IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev 223:20–38. https://doi.org/10.1111/j.1600-065X.2008.00624.x
McAfoose J, Baune BT (2009) Evidence for a cytokine model of cognitive function. Neurosci Biobehav Rev 33(3):355–366. https://doi.org/10.1016/j.neubiorev.2008.10.005
Zhang Y, Liu L, Liu YZ, Shen XL, Wu TY, Zhang T, Wang W, Wang YX, Jiang CL (2015) NLRP3 inflammasome mediates chronic mild stress-induced depression in mice via neuroinflammation. Int J Neuropsychopharmacol 18(8). https://doi.org/10.1093/ijnp/pyv006
Kim HK, Chen W, Andreazza AC (2015) The potential role of the NLRP3 inflammasome as a link between mitochondrial complex I dysfunction and inflammation in bipolar disorder. Neural Plast 2015:10. https://doi.org/10.1155/2015/408136
Kim HK, Andreazza AC, Elmi N, Chen W, Young LT (2016) Nod-like receptor pyrin containing 3 (NLRP3) in the post-mortem frontal cortex from patients with bipolar disorder: a potential mediator between mitochondria and immune-activation. J Psychiatr Res 72:43–50. https://doi.org/10.1016/j.jpsychires.2015.10.015
Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T, Rouis M (2015) NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol 4:296–307. https://doi.org/10.1016/j.redox.2015.01.008
Rodríguez-Muela N, Hernández-Pinto AM, Serrano-Puebla A, García-Ledo L, Latorre SH, de la Rosa EJ, Boya P (2015) Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ 22(3):476–487. https://doi.org/10.1038/cdd.2014.203
Boya P, Kroemer G (2008) Lysosomal membrane permeabilization in cell death. Oncogene 27(50):6434–6451. https://doi.org/10.1038/onc.2008.310
Kreuzaler PA, Staniszewska AD, Li W, Omidvar N, Kedjouar B, Turkson J, Poli V, Flavell RA et al (2011) Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol 13(3):303–309. https://doi.org/10.1038/ncb2171
Kurz T, Leake A, von Zglinicki T, Brunk UT (2004) Lysosomal redox-active iron is important for oxidative stress-induced DNA damage. Ann N Y Acad Sci 1019:285–288. https://doi.org/10.1196/annals.1297.048
Persson HL, Yu Z, Tirosh O, Eaton JW, Brunk UT (2003) Prevention of oxidant-induced cell death by lysosomotropic iron chelators. Free Radic Biol Med 34(10):1295–1305
Zhang L, Sheng R, Qin Z (2009) The lysosome and neurodegenerative diseases. Acta Biochim Biophys Sin 41(6):437–445
Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, Burrage AM, Sudholt S et al (2013) Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One 8(4):e62143. https://doi.org/10.1371/journal.pone.0062143
Terman A, Kurz T, Gustafsson B, Brunk UT (2006) Lysosomal labilization. IUBMB Life 58(9):531–539. https://doi.org/10.1080/15216540600904885
Villalpando Rodriguez GE, Torriglia A (2013) Calpain 1 induce lysosomal permeabilization by cleavage of lysosomal associated membrane protein 2. Biochim Biophys Acta 1833(10):2244–2253. https://doi.org/10.1016/j.bbamcr.2013.05.019
Yamashima T, Oikawa S (2009) The role of lysosomal rupture in neuronal death. Prog Neurobiol 89(4):343–358. https://doi.org/10.1016/j.pneurobio.2009.09.003
Usenovic M, Krainc D (2012) Lysosomal dysfunction in neurodegeneration: the role of ATP13A2/PARK9. Autophagy 8(6):987–988. https://doi.org/10.4161/auto.20256
Machado-Vieira R, Andreazza AC, Viale CI, Zanatto V, Cereser V, Vargas RS, Kapczinski F, Portela LV et al (2007) Oxidative stress parameters in unmedicated and treated bipolar subjects during initial manic episode: a possible role for lithium antioxidant effects. Neurosci Lett 421(1):33–36. https://doi.org/10.1016/j.neulet.2007.05.016
Pirooznia M, Wang T, Avramopoulos D, Potash JB, Zandi PP, Goes FS (2016) High-throughput sequencing of the synaptome in major depressive disorder. Mol Psychiatry 21(5):650–655. https://doi.org/10.1038/mp.2015.98
Davidovich P, Kearney CJ, Martin SJ (2014) Inflammatory outcomes of apoptosis, necrosis and necroptosis. Biol Chem 395(10):1163–1171. https://doi.org/10.1515/hsz-2014-0164
Challa S, Chan FK (2010) Going up in flames: necrotic cell injury and inflammatory diseases. Cell Mol Life Sci: CMLS 67(19):3241–3253. https://doi.org/10.1007/s00018-010-0413-8
Lucas K, Maes M (2013) Role of the Toll like receptor (TLR) radical cycle in chronic inflammation: possible treatments targeting the TLR4 pathway. Mol Neurobiol 48(1):190–204. https://doi.org/10.1007/s12035-013-8425-7
Lucas K, Morris G, Anderson G, Maes M (2015) The Toll-like receptor radical cycle pathway: a new drug target in immune-related chronic fatigue. CNS Neurol Disord Drug Targets 14(7):838–854
Land WG (2015) The role of damage-associated molecular patterns in human diseases: part I—promoting inflammation and immunity. Sultan Qaboos Univ Med J 15(1):e9–e21
Garcia-Martinez I, Shaker ME, Mehal WZ (2015) Therapeutic opportunities in damage-associated molecular pattern-driven metabolic diseases. Antioxid Redox Signal 23(17):1305–1315. https://doi.org/10.1089/ars.2015.6383
Thundyil J, Lim KL (2015) DAMPs and neurodegeneration. Ageing Res Rev 24(Pt A):17–28. https://doi.org/10.1016/j.arr.2014.11.003
Wilkins HM, Weidling IW, Ji Y, Swerdlow RH (2017) Mitochondria-derived damage-associated molecular patterns in neurodegeneration. Front Immunol 8:508. https://doi.org/10.3389/fimmu.2017.00508
Stertz L, Fries GR, Rosa AR, Kauer-Sant'anna M, Ferrari P, Paz AV, Green C, Cunha AB et al (2015) Damage-associated molecular patterns and immune activation in bipolar disorder. Acta Psychiatr Scand 132(3):211–217. https://doi.org/10.1111/acps.12417
Sorci G, Bianchi R, Riuzzi F, Tubaro C, Arcuri C, Giambanco I, Donato R (2010) S100B protein, a damage-associated molecular pattern protein in the brain and heart, and beyond. Cardiovasc Psychiatry Neurol 2010:656481. https://doi.org/10.1155/2010/656481
Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, Ye J, Zhang X et al (2015) Activation of necroptosis in multiple sclerosis. Cell Rep 10(11):1836–1849. https://doi.org/10.1016/j.celrep.2015.02.051
Zhou W, Yuan J (2014) Necroptosis in health and diseases. Semin Cell Dev Biol 35:14–23. https://doi.org/10.1016/j.semcdb.2014.07.013
Belaidi AA, Bush AI (2016) Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J Neurochem 139(Suppl 1):179–197. https://doi.org/10.1111/jnc.13425
Guiney SJ, Adlard PA, Bush AI, Finkelstein DI, Ayton S (2017) Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem Int 104:34–48. https://doi.org/10.1016/j.neuint.2017.01.004
Do Van B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, Bastide M, Laloux C et al (2016) Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis 94:169–178. https://doi.org/10.1016/j.nbd.2016.05.011
Andreazza AC, Gildengers A, Rajji TK, Zuzarte PML, Mulsant BH, Young LT (2015) Oxidative stress in older patients with bipolar disorder. Am J Geriatr psychiatry: Off J Am Assoc Geriatr Psychiatry 23(3):314–319. https://doi.org/10.1016/j.jagp.2014.05.008
Wang JF, Shao L, Sun X, Young LT (2009) Increased oxidative stress in the anterior cingulate cortex of subjects with bipolar disorder and schizophrenia. Bipolar Disord 11(5):523–529. https://doi.org/10.1111/j.1399-5618.2009.00717.x
Medina-Hernandez V, Ramos-Loyo J, Luquin S, Sanchez LF, Garcia-Estrada J, Navarro-Ruiz A (2007) Increased lipid peroxidation and neuron specific enolase in treatment refractory schizophrenics. J Psychiatr Res 41(8):652–658. https://doi.org/10.1016/j.jpsychires.2006.02.010
Ramos-Loyo J, Medina-Hernández V, Estarrón-Espinosa M, Canales-Aguirre A, Gómez-Pinedo U, Cerdán-Sánchez LF (2013) Sex differences in lipid peroxidation and fatty acid levels in recent onset schizophrenia. Prog Neuro-Psychopharmacol Biol Psychiatry 44:154–161. https://doi.org/10.1016/j.pnpbp.2013.02.007
Selley ML (2004) Increased (E)-4-hydroxy-2-nonenal and asymmetric dimethylarginine concentrations and decreased nitric oxide concentrations in the plasma of patients with major depression. J Affect Disord 80(2–3):249–256. https://doi.org/10.1016/s0165-0327(03)00135-6
Romano A, Serviddio G, Calcagnini S, Villani R, Giudetti AM, Cassano T, Gaetani S Linking lipid peroxidation and neuropsychiatric disorders: focus on 4-hydroxy-2-nonenal. Free Radic Biol Med. https://doi.org/10.1016/j.freeradbiomed.2016.12.046
Gaschler MM, Stockwell BR (2017) Lipid peroxidation in cell death. Biochem Biophys Res Commun 482(3):419–425. https://doi.org/10.1016/j.bbrc.2016.10.086
Shichiri M (2014) The role of lipid peroxidation in neurological disorders. J Clin Biochem Nutr 54(3):151–160. https://doi.org/10.3164/jcbn.14-10
Choi Y, Kim HS, Shin KY, Kim EM, Kim M, Kim HS, Park CH, Jeong YH et al (2007) Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer’s disease models. Neuropsychopharmacol: Off Publ Am Coll Neuropsychopharmacol 32(11):2393–2404. https://doi.org/10.1038/sj.npp.1301377
Abdel-Salam OM (2008) Drugs used to treat Parkinson’s disease, present status and future directions. CNS Neurol Disord Drug Targets 7(4):321–342
Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS et al (2002) Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 417(6884):74–78. https://doi.org/10.1038/417074a
Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L et al (2000) Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 6(7):797–801. https://doi.org/10.1038/77528
Brundula V, Rewcastle NB, Metz LM, Bernard CC, Yong VW (2002) Targeting leukocyte MMPs and transmigration: minocycline as a potential therapy for multiple sclerosis. Brain 125(Pt 6):1297–1308
Nessler S, Dodel R, Bittner A, Reuss S, Du Y, Hemmer B, Sommer N (2002) Effect of minocycline in experimental autoimmune encephalomyelitis. Ann Neurol 52(5):689–690; author reply 690. https://doi.org/10.1002/ana.10353
Garrido-Mesa N, Zarzuelo A, Gálvez J (2013) Minocycline: far beyond an antibiotic. Br J Pharmacol 169(2):337–352. https://doi.org/10.1111/bph.12139
Stirling DP, Koochesfahani KM, Steeves JD, Tetzlaff W (2005) Minocycline as a neuroprotective agent. Neuroscientist: Rev J Bringing Neurobiol, Neurol Psychiatry 11(4):308–322. https://doi.org/10.1177/1073858405275175
Kim HS, Suh YH (2009) Minocycline and neurodegenerative diseases. Behav Brain Res 196(2):168–179. https://doi.org/10.1016/j.bbr.2008.09.040
Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, Sheridan JF, Godbout JP (2008) Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflammation 5:15. https://doi.org/10.1186/1742-2094-5-15
Seabrook TJ, Jiang L, Maier M, Lemere CA (2006) Minocycline affects microglia activation, Abeta deposition, and behavior in APP-tg mice. Glia 53(7):776–782. https://doi.org/10.1002/glia.20338
Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H et al (2002) Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22(5):1763–1771
Koistinaho M, Malm TM, Kettunen MI, Goldsteins G, Starckx S, Kauppinen RA, Opdenakker G, Koistinaho J (2005) Minocycline protects against permanent cerebral ischemia in wild type but not in matrix metalloprotease-9-deficient mice. J Cereb Blood Flow Metab 25(4):460–467. https://doi.org/10.1038/sj.jcbfm.9600040
Festoff BW, Ameenuddin S, Arnold PM, Wong A, Santacruz KS, Citron BA (2006) Minocycline neuroprotects, reduces microgliosis, and inhibits caspase protease expression early after spinal cord injury. J Neurochem 97(5):1314–1326. https://doi.org/10.1111/j.1471-4159.2006.03799.x
Antonenko YN, Rokitskaya TI, Cooper AJ, Krasnikov BF (2010) Minocycline chelates Ca2+, binds to membranes, and depolarizes mitochondria by formation of Ca2+-dependent ion channels. J Bioenerg Biomembr 42(2):151–163. https://doi.org/10.1007/s10863-010-9271-1
Garcia-Martinez EM, Sanz-Blasco S, Karachitos A, Bandez MJ, Fernandez-Gomez FJ, Perez-Alvarez S, de Mera RM, Jordan MJ et al (2010) Mitochondria and calcium flux as targets of neuroprotection caused by minocycline in cerebellar granule cells. Biochem Pharmacol 79(2):239–250. https://doi.org/10.1016/j.bcp.2009.07.028
Scarabelli TM, Stephanou A, Pasini E, Gitti G, Townsend P, Lawrence K, Chen-Scarabelli C, Saravolatz L et al (2004) Minocycline inhibits caspase activation and reactivation, increases the ratio of XIAP to smac/DIABLO, and reduces the mitochondrial leakage of cytochrome C and smac/DIABLO. J Am Coll Cardiol 43(5):865–874. https://doi.org/10.1016/j.jacc.2003.09.050
Matsukawa N, Yasuhara T, Hara K, Xu L, Maki M, Yu G, Kaneko Y, Ojika K et al (2009) Therapeutic targets and limits of minocycline neuroprotection in experimental ischemic stroke. BMC Neurosci 10:126. https://doi.org/10.1186/1471-2202-10-126
Li J, Chen J, Mo H, Chen J, Qian C, Yan F, Gu C, Hu Q et al (2016) Minocycline protects against NLRP3 inflammasome-induced inflammation and P53-associated apoptosis in early brain injury after subarachnoid hemorrhage. Mol Neurobiol 53(4):2668–2678. https://doi.org/10.1007/s12035-015-9318-8
Sinha-Hikim I, Shen R, Nzenwa I, Gelfand R, Mahata SK, Sinha-Hikim AP (2011) Minocycline suppresses oxidative stress and attenuates fetal cardiac myocyte apoptosis triggered by in utero cocaine exposure. Apoptosis: Int J Program Cell Death 16(6):563–573. https://doi.org/10.1007/s10495-011-0590-4
Blum D, Chtarto A, Tenenbaum L, Brotchi J, Levivier M (2004) Clinical potential of minocycline for neurodegenerative disorders. Neurobiol Dis 17(3):359–366. https://doi.org/10.1016/j.nbd.2004.07.012
Domercq M, Matute C (2004) Neuroprotection by tetracyclines. Trends Pharmacol Sci 25(12):609–612. https://doi.org/10.1016/j.tips.2004.10.001
Tao R, Kim SH, Honbo N, Karliner JS, Alano CC (2010) Minocycline protects cardiac myocytes against simulated ischemia-reperfusion injury by inhibiting poly(ADP-ribose) polymerase-1. J Cardiovasc Pharmacol 56(6):659–668. https://doi.org/10.1097/FJC.0b013e3181faeaf0
Schonfeld P, Siemen D, Kreutzmann P, Franz C, Wojtczak L (2013) Interaction of the antibiotic minocycline with liver mitochondria—role of membrane permeabilization in the impairment of respiration. FEBS J 280(24):6589–6599. https://doi.org/10.1111/febs.12563
Cuenca-Lopez MD, Karachitos A, Massarotto L, Oliveira PJ, Aguirre N, Galindo MF, Kmita H, Jordan J (2012) Minocycline exerts uncoupling and inhibiting effects on mitochondrial respiration through adenine nucleotide translocase inhibition. Pharmacol Res 65(1):120–128. https://doi.org/10.1016/j.phrs.2011.08.007
Alano CC, Kauppinen TM, Valls AV, Swanson RA (2006) Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proc Natl Acad Sci U S A 103(25):9685–9690. https://doi.org/10.1073/pnas.0600554103
Stirling DP, Khodarahmi K, Liu J, McPhail LT, McBride CB, Steeves JD, Ramer MS, Tetzlaff W (2004) Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci 24(9):2182–2190. https://doi.org/10.1523/JNEUROSCI.5275-03.2004
Yong VW, Wells J, Giuliani F, Casha S, Power C, Metz LM (2004) The promise of minocycline in neurology. Lancet Neurol 3(12):744–751. https://doi.org/10.1016/S1474-4422(04)00937-8
Maier K, Merkler D, Gerber J, Taheri N, Kuhnert AV, Williams SK, Neusch C, Bahr M et al (2007) Multiple neuroprotective mechanisms of minocycline in autoimmune CNS inflammation. Neurobiol Dis 25(3):514–525. https://doi.org/10.1016/j.nbd.2006.10.022
Shahzad K, Bock F, Al-Dabet MM, Gadi I, Nazir S, Wang H, Kohli S, Ranjan S et al (2016) Stabilization of endogenous Nrf2 by minocycline protects against Nlrp3-inflammasome induced diabetic nephropathy. Sci Rep 6:34228. https://doi.org/10.1038/srep34228
Sonmez E, Kabatas S, Ozen O, Karabay G, Turkoglu S, Ogus E, Yilmaz C, Caner H et al (2013) Minocycline treatment inhibits lipid peroxidation, preserves spinal cord ultrastructure, and improves functional outcome after traumatic spinal cord injury in the rat. Spine 38(15):1253–1259. https://doi.org/10.1097/BRS.0b013e3182895587
Homsi S, Federico F, Croci N, Palmier B, Plotkine M, Marchand-Leroux C, Jafarian-Tehrani M (2009) Minocycline effects on cerebral edema: relations with inflammatory and oxidative stress markers following traumatic brain injury in mice. Brain Res 1291:122–132. https://doi.org/10.1016/j.brainres.2009.07.031
Zhao F, Hua Y, He Y, Keep RF, Xi G (2011) Minocycline-induced attenuation of iron overload and brain injury following experimental intracerebral hemorrhage. Stroke 42(12):3587–3593. https://doi.org/10.1161/STROKEAHA.111.623926
Zhao F, Xi G, Liu W, Keep RF, Hua Y (2016) Minocycline attenuates iron-induced brain injury. Acta Neurochir Suppl 121:361–365. https://doi.org/10.1007/978-3-319-18497-5_62
O'Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, Kelley KW, Dantzer R (2009) Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry 14(5):511–522. https://doi.org/10.1038/sj.mp.4002148
Dean OM, Kanchanatawan B, Ashton M, Mohebbi M, Ng CH, Maes M, Berk L, Sughondhabirom A et al (2017) Adjunctive minocycline treatment for major depressive disorder: a proof of concept trial. Aust N Z J Psychiatry 51(8):829–840. https://doi.org/10.1177/0004867417709357
Berk M, Dean OM, Cotton SM, Jeavons S, Tanious M, Kohlmann K, Hewitt K, Moss K et al (2014) The efficacy of adjunctive N-acetylcysteine in major depressive disorder: a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry 75(6):628–636. https://doi.org/10.4088/JCP.13m08454
Abdel Baki SG, Schwab B, Haber M, Fenton AA, Bergold PJ (2010) Minocycline synergizes with N-acetylcysteine and improves cognition and memory following traumatic brain injury in rats. PLoS One 5(8):e12490. https://doi.org/10.1371/journal.pone.0012490
Dean OM, van den Buuse M, Berk M, Copolov DL, Mavros C, Bush AI (2011) N-acetyl cysteine restores brain glutathione loss in combined 2-cyclohexene-1-one and d-amphetamine-treated rats: relevance to schizophrenia and bipolar disorder. Neurosci Lett 499(3):149–153. https://doi.org/10.1016/j.neulet.2011.05.027
Dean O, Giorlando F, Berk M (2011) N-acetylcysteine in psychiatry: current therapeutic evidence and potential mechanisms of action. J Psychiatry Neurosci: JPN 36(2):78–86. https://doi.org/10.1503/jpn.100057
Berk M, Malhi GS, Gray LJ, Dean OM (2013) The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol Sci 34(3):167–177. https://doi.org/10.1016/j.tips.2013.01.001
Tai IH, Sheen JM, Lin YJ, Yu HR, Tiao MM, Chen CC, Huang LT, Tain YL (2016) Maternal N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and prevents programmed hypertension in male offspring exposed to prenatal dexamethasone and postnatal high-fat diet. Nitric Oxide: Biol Chem / Off J Nitric Oxide Soc 53:6–12. https://doi.org/10.1016/j.niox.2015.12.006
Halasi M, Wang M, Chavan TS, Gaponenko V, Hay N, Gartel AL (2013) ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem J 454(2):201–208. https://doi.org/10.1042/bj20130282
Zhang F, Lau SS, Monks TJ (2011) The cytoprotective effect of N-acetyl-L-cysteine against ROS-induced cytotoxicity is independent of its ability to enhance glutathione synthesis. Toxicol Sci 120(1):87–97. https://doi.org/10.1093/toxsci/kfq364
Jiao Y, Ma S, Wang Y, Li J, Shan L, Liu Q, Liu Y, Song Q et al (2016) N-acetyl cysteine depletes reactive oxygen species and prevents dental monomer-induced intrinsic mitochondrial apoptosis in vitro in human dental pulp cells. PLoS One 11(1):e0147858. https://doi.org/10.1371/journal.pone.0147858
Kose SA, Naziroglu M (2015) N-acetyl cysteine reduces oxidative toxicity, apoptosis, and calcium entry through TRPV1 channels in the neutrophils of patients with polycystic ovary syndrome. Free Radic Res 49(3):338–346. https://doi.org/10.3109/10715762.2015.1006214
Baker DA, Madayag A, Kristiansen LV, Meador-Woodruff JH, Haroutunian V, Raju I (2008) Contribution of cystine-glutamate antiporters to the psychotomimetic effects of phencyclidine. Neuropsychopharmacol: Off Publ Am Coll Neuropsychopharmacol 33(7):1760–1772. https://doi.org/10.1038/sj.npp.1301532
Kau KS, Madayag A, Mantsch JR, Grier MD, Abdulhameed O, Baker DA (2008) Blunted cystine-glutamate antiporter function in the nucleus accumbens promotes cocaine-induced drug seeking. Neuroscience 155(2):530–537. https://doi.org/10.1016/j.neuroscience.2008.06.010
Kupchik YM, Moussawi K, Tang XC, Wang X, Kalivas BC, Kolokithas R, Ogburn KB, Kalivas PW (2012) The effect of N-acetylcysteine in the nucleus accumbens on neurotransmission and relapse to cocaine. Biol Psychiatry 71(11):978–986. https://doi.org/10.1016/j.biopsych.2011.10.024
Bridges RJ, Natale NR, Patel SA (2012) System x(c)(−) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol 165(1):20–34. https://doi.org/10.1111/j.1476-5381.2011.01480.x
Madayag A, Lobner D, Kau KS, Mantsch JR, Abdulhameed O, Hearing M, Grier MD, Baker DA (2007) Repeated N-acetylcysteine administration alters plasticity-dependent effects of cocaine. J Neurosci 27(51):13968–13976. https://doi.org/10.1523/jneurosci.2808-07.2007
Deepmala SJ, Kumar N, Delhey L, Berk M, Dean O, Spielholz C, Frye R (2015) Clinical trials of N-acetylcysteine in psychiatry and neurology: a systematic review. Neurosci Biobehav Rev 55:294–321. https://doi.org/10.1016/j.neubiorev.2015.04.015
Minarini A, Ferrari S, Galletti M, Giambalvo N, Perrone D, Rioli G, Galeazzi GM (2017) N-acetylcysteine in the treatment of psychiatric disorders: current status and future prospects. Expert Opin Drug Metab Toxicol 13(3):279–292. https://doi.org/10.1080/17425255.2017.1251580
Funding
MB is supported by a NHMRC Senior Principal Research Fellowship 1059660.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
MB has received Grant/Research Support from the NIH, Cooperative Research Centre, Simons Autism Foundation, Cancer Council of Victoria, Stanley Medical Research Foundation, MBF, NHMRC, Beyond Blue, Rotary Health, Geelong Medical Research Foundation, Bristol Myers Squibb, Eli Lilly, Glaxo SmithKline, Meat and Livestock Board, Organon, Novartis, Mayne Pharma, Servier and Woolworths; has been a speaker for Astra Zeneca, Bristol Myers Squibb, Eli Lilly, Glaxo SmithKline, Janssen Cilag, Lundbeck, Merck, Pfizer, Sanofi Synthelabo, Servier, Solvay and Wyeth; and served as a consultant to Astra Zeneca, Bioadvantex, Bristol Myers Squibb, Eli Lilly, Glaxo SmithKline, Janssen Cilag, Lundbeck Merck and Servier.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Morris, G., Walker, A.J., Berk, M. et al. Cell Death Pathways: a Novel Therapeutic Approach for Neuroscientists. Mol Neurobiol 55, 5767–5786 (2018). https://doi.org/10.1007/s12035-017-0793-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0793-y