
Abstract
A phenoxy-imine proligand with the additional OH donor group, 4,6-tBu2-2-(2-CH2(OH)-C6H4N = CH)C6H3OH (LH2), was synthesized and used to prepare group 4 and 5 complexes by reacting with Ti(OiPr)4 (LTi) and VO(OiPr)3 (LV). All new compounds were characterized by the FTIR, 1H and 13C NMR spectroscopy and LTi by the single-crystal X-ray diffraction analysis. The complexes were used as catalysts in the ring opening polymerization of ε-caprolactone. The influence of monomer/transition metal molar ratio, reaction time, polymerization temperature as well as complex type was investigated in detail. The complexes showed high (LTi) and moderate (LV) activity in ε-caprolactone polymerization and the resultant polycaprolactones exhibited Mn and Mw/Mn values ranging from 4.0 · 103 to 18.7 · 103 g/mol and from 1.4 to 2.5, respectively.
Similar content being viewed by others

Introduction
Poly(ε-caprolactone) (PCL) belongs to the group of biodegradable polymers. Those materials have attracted a great interest in both biomedical and traditional applications [1,2,3,4,5,6] due to good mechanical strength, biocompatibility, bioresorbability, compatibility with a large range of other polymers and notable rheological and viscoelastic properties. PCL can be synthesized by different mechanisms. However, the ring opening polymerization of ε-caprolactone (CL) in the presence of well-defined metal catalysts seems to be the most convenient to prepare this polymer [3, 7, 8]. Complexes which are based on metals with empty p, d or f orbitals polymerize CL via the coordination/insertion mechanism and they often offer good control over the polymer molar mass and provide polymers with narrow dispersity and with well-defined end groups [9]. Many various metal complexes were explored for PCL synthesis [9], including aluminium [10,11,12,13,14,15,16,17,18,19,20], magnesium [10, 13, 21], zinc [8, 10, 20, 22, 23] and rare earth metal complexes [9, 24, 25]. Transition metal complexes also drew up the attention of researchers in recent years, and typically titanium [20, 26,27,28,29,30,31,32,33,34,35,36,37] and zirconium [29, 33, 35, 38] ones. These well-defined complexes are usually supported by bi- or higher dentate ligands which offer the opportunity of adjusting the steric and electronic effect of the ligand skeleton. Bis(phenolate) [26, 28], amine-bis(phenolate) [27], diamine-bis(phenolate) [29, 39], 2,6-bis(hydroxyalkyl)pyridine [30], bis(sulfonamide)amine [32], aminodiol [34] and Schiff base type compounds [31, 33, 35, 38] can be listed as good examples. The last group is one of the most versatile and easily obtainable group of ancillary ligands [40].
The complexes with ONNO-tetradentate ligands of Schiff base type, and namely dinuclear salen complexes of Zr and Hf, polymerize CL with good efficiency and with a great degree of molar mass control under solvent-free conditions [38]. High activity and control of CL polymerization was shown by the complexes of group 4 metals, especially those bearing ONN-tridentate phenoxy-imine ligands derived from 8-aminoquinoline or those bearing ONO-tridentate Schiff base ligands derived from o-vanillin [31, 35]. The steric bulkiness of the imine nitrogen substituents in the latter influenced the ligand coordination modes in the complexes and hence their catalytic activity [31]. The complexes of Ti with ONO-tridentate Schiff bases, as obtained from 2-hydroxybenzaldehyde derivatives, were found to be more catalytically active than other previously investigated Ti complexes which were bearing multi-dentate phenolate ligands. Authors suggested that such ligands reduces the occupied space and increases the monomer coordination around the Ti center, thus improve catalytic activity [33]. It was also disclosed that the electron-donating substituents of the ligands did not enhance the catalytic activity in CL polymerization [33]. These research inspired us to prepare new, potentially highly active, titanium complex bearing simple tridentate ONO-Schiff base ligand based on 2-[{2-(hydroxymethyl)phenylimino}-methyl]-4,6-di-tert-butylophenol. Furthermore, it is known that the vanadium complexes are definitely less often used in the CL polymerization reaction as compared to group 4 complexes [41,42,43,44], especially those based on a Schiff base ligand [42]. After the screening tests, the vanadium(III) phenoxy-imine complexes supported by ON-bidentate ligands and related tetradentate Schiff base ligands, in the presence of benzyl alcohol as a co-initiator, were found to produce low molar mass products of low dispersity [42]. Therefore the vanadium complex based on synthesized tridentate phenoxy-imine ligand was also synthesized and both complexes were examined in the CL polymerization process. Titanium complex was also tested in polymerization of racemic lactide.
Experimental section
Chemicals and reagents
Argon (grade 5.0, Linde Gas), Ti(OiPr)4 (99,999%, Sigma-Aldrich), VO(OiPr)3 (Sigma-Aldrich), methanol (Stanlab, analytical grade), 3,5-di-tert-butyl-2-hydroxybenzaldehyde (99%, Sigma-Aldrich), 2-aminobenzyl alcohol (98%, Sigma-Aldrich) were used as received. CDCl3 (99,5 atom % D, Deutero GmbH or 99,8 atom % D, 0.03% TMS), CH2Cl2 (Chempur, analytical grade), ε-caprolactone (99%, Acros Organic) and L,D-lactide (99%, Acros Organic) were dried over molecular sieves. Hexane (Stanlab, analytical grade) and toluene (Chempur, ACS grade) were refluxed over sodium and sodium/benzophenone, respectively and distilled under nitrogen atmosphere prior to use.
Methods
All manipulations of air and/or moisture sensitive materials were performed under an argon atmosphere using dual vacuum/argon line and Schlenk-type glassware. The infrared spectra were recorded with the Nicole Nexus 2002 FTIR spectrometer. The samples of ligand and complexes were prepared in nujol under inert atmosphere and their scans were taken in the range from 4000 to 400 cm−1 with a 2 cm−1 resolution. 1H and 13C NMR spectra were taken on the Bruker Ultrashield 400 spectrometer at room temperature using CDCl3 as a solvent. Chemical shifts were reported in ppm and were referenced to solvent residue peak at 7.26 ppm and 77.16 ppm in the proton and carbon spectra, respectively. Differential scanning calorimetry (DSC) thermograms were acquired with a 2010 DSC calorimeter from TA Instruments at the heating rate 10 °C/min. The heating–cooling-heating cycles were performed and the melting temperatures (Tm) and crystallinities (χ) of polymers were obtained from the second heating. The poly(ε-caprolactone) crystallinity was calculated assuming that the enthalpy of melting of completely crystalline polymer is equal to 139.5 J/g [1]. Molar mass and dispersity of polymers were measured by size exclusion chromatography (SEC) using detector Waters 2414 RI and three styragel columns (HR1, HR2 and HR4). Analyses were made at 35 °C using THF as eluting solvent under a flow rate of 0.6 mL/min. Polystyrene standards (Shodex) were used for calibration. Obtained Mn values were multiplied by 0.56 (correction coefficient) to correlate them to actual values (Mn(corr)) [45].
X-ray crystallography
The single crystals of the LTi has been mounted on a CCD Xcalibur diffractometer (graphite monochromatic, MoKα radiation, λ = 0.71073 Å) at 100.0(1) K. The reciprocal space was explored by ω scans with detector positions at 60 mm distance from the crystal. The diffraction data processing of studied compounds (Lorentz and polarization corrections were applied) was performed using the CrysAlis CCD [46]. The structure was solved in the triclinic crystal system, P‾1 space group (Table S1), by direct methods and refined by a full-matrix least-squares method using SHELXL14 program [47, 48]. The H atoms were located from difference Fourier synthesis and from geometrical parameters and refined using a riding model. The structure drawings were prepared using SHELXTL and Mercury [49] programs. Crystallographic data for solved structures have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC: 1,877,301. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: 144 1223 336 033; email:deposit@ccdc.cam.ac.uk.
Synthesis of 2-[{2-(hydroxymethyl)phenylimino}-methyl]-4,6-di-tert-butylophenol (LH2)
2-aminobenzyl alcohol (1.29 g, 0.01 mol) and 3,5-di-tert-butyl-2-hydroxybenzaldehyde (2.34 g, 0.01 mol) were dissolved in ethanol and heated to reflux. The reactions were carried out for 48 h. The solvent was evaporated and the oily residue was purified by flash chromatography to give 2.33 g of product. Yield: 69%. 1H NMR (400 MHz, CDCl3): δ 1.34 (s, 9H, tBu), 1.48 (s, 9H, tBu); 4.90 (s, 2H, Ar-CH2-OH); 7.13 (dd, J = 7,8; 1.0 Hz, 1H, Ar–H), 7.24 (d, J = 2.4 Hz, 1H, Ar–H), 7.30 (td, J = 7.4; 1,2 Hz, 1H, Ar–H), 7.37 (td, J = 7.6; 1.5 Hz, 1H, Ar–H), 7.48 (d, J = 2.4 Hz, 1H, Ar–H), 7.53 (dd, J = 7.3; 0.9 Hz, 1H, Ar–H); 8.62 (s, 1H, HC = N); 13.39 (s, 1H, Ar-OH). 13C NMR (100.6 MHz, CDCl3): δ 29.54 (3C, tBu), 31.60 (3C, tBu), 34.34 (1C, tBu), 35.28 (1C, tBu); 62.13 (1C, Ar-CH2-OH); 118.23, 118.54, 126.92, 127.11, 128.24, 128.52, 128.97, 134.54, 137.25, 140.92, 147.10 (Ar), 158.34 (1C, Ar-OH); 164.76 (1C, HC = N). FTIR (nujol, cm−1): 3311 ν(O–H), 1616 ν(C = N).
Synthesis of complexes
Complex LTi. A solution of titanium(IV) isopropoxide (0.42 mL, 1.414 mmol) in 5 mL of toluene was added dropwise into an orange solution of LH2 (0.480 g, 1.414 mmol) in 15 mL of toluene over 15 min. The clear solution after overnight stirring at room temperature was evaporated to dryness and the yellow residue was dissolved in 5 mL of dry methylene chloride. Solvent evaporation under vacuum afforded yellow-orange powder. Yield: 0.490 g, 68.9%. 1H NMR (400 MHz, CDCl3): δ 0.52 (d, 1H, J = 6.0 Hz, O–CH(CH3)2), 0.63 (d, 1 H, J = 6.1 Hz, O–CH(CH3)2), 0.76 (d, 1H, J = 6.0 Hz, O–CH(CH3)2), 1.21 (d, 1,H, J = 6.1 Hz, O–CH(CH3)2), 0.96, 1.04, 1.05, 1.07, 1.30, 1.34, 1.40, 1.42, 1.49, 1.51 (26 H, tBu and O–CH(CH3)2, overlaping; 4.65 (m, 1H, O–CH(CH3)2); 4.52 (m, 0.4 H, O–CH(CH3)2); 4.03 and 3.97 (overlapping multiplets, 0.6 H, O–CH(CH3)2); 4.93 (s, 0.6 H, Ar-CH2-O); 5.09 (s, 1 H, Ar-CH2-O), 5.12 (s, 0.2 H, Ar-CH2-O); 5.17 (s, 0.2 H, Ar-CH2-O); 6.80 (d, 0.5H, J = 7.4 Hz); 6.94 (d, 0.5H, J = 7.6 Hz); 7.41—7.06 (4H, Ar–H), 7.57 (dd, J = 8.5 Hz, 2.1 Hz, 1H); 8.42, 8.43, 8.47 (1H, HC = N). 13C NMR (100.6 MHz, CDCl3): δ 25.53, 25.94, 25.99, 27.07, 29.26, 29.66, 29.84, 30.14, 31.51, 31.57, 31.73, 34.25, 34.33, 34.39, 34.88, 35.51, 64.63, 73.23, 73,51, 77.96, 120.71, 122.38, 125.44, 126.39, 126.64, 126.69, 126.87, 126.95, 128.39, 128.73, 128.96, 130.32, 131.04, 131.34, 132.6, 149.90, 161.77, 164.26, 165.79, 166.37. FTIR (nujol, cm−1) 1611 ν(C = N). The single crystals of LTi which were suitable for X-ray analysis were grown from toluene in a freezer.
Complex LV. A solution of vanadium(V) oxytriisopropoxide (0.235 mL, 1.084 mmol) in 10 mL of toluene was added dropwise to a stirred orange solution of LH2 (0.368 g, 1.084 mmol) in 10 mL of toluene. The reaction solution was stirred at room temperature overnight and then it was concentrated in vacuo to 2 mL. To the residue, 5 mL of CH2Cl2 was added and after stirring for 30 min solvent was removed under vacuum. The operation was repeated with 20 mL of hexane. Solvent evaporation under vacuum afforded dark green powder. Yield: 0.439 g, 87.4%. 1H NMR (400 MHz, CDCl3): main isomer 1.42 (d, 3H, J = 6.1 Hz, O–CH(CH3)2), 1.50 (d, 3H, J = 6.1 Hz, O–CH(CH3)2); 1.35 (s, 9H, tBu); 1.49 (s, 9H, tBu); 5.44 (d, 1H, J = 13.9 Hz, Ar-CH2-O), 5.67 (d, 1H, J = 13.7 Hz, Ar-CH2-O); 5.79–5.86 (m, 1H, O–CH(CH3)2); 7.10 (dd, 0.2 H, J = 7.6 Hz, Ar–H); 7.19 (dd, 0.8 H, J = 7.6 Hz, Ar–H), 7.32 (d, 1H, J = 2.5 Hz, Ar–H), 7.33–7.42 (m, 3H, Ar–H), 7.64 (d, 1H, J = 2.5 Hz, Ar–H); 8.53 (s, 1H, CH = N). 13C NMR (100.6 MHz, CDCl3): δ 25.55, 25.78, 29.45, 29.73, 29.98, 31.55, 31.87, 35.29, 35.53, 64.64, 120.98, 121,46, 122,13, 125.13, 126.90, 127.58, 128.04, 129.22, 147.04, 156.40, 163.27, 168.66. FTIR (nujol, cm−1) 1612 ν(C = N).
Polymerization of ε-caprolactone
For the bulk polymerization tests, about 20–30 mg of a complex and the appropriate amount of ε-caprolactone (resulting from the assumed monomer/transition metal molar ratio) were put into a Schlenk tube at room temperature under argon. The tube was then placed on the oil bath which had been pre-heated to 120 °C (or to other polymerization temperature, as chosen for a test) and the mixture was stirred at that temperature for a specified amount of time. After the reaction was quenched with the use of methanol, the polymer was extracted with chloroform and precipitated in methanol. The polymer was separated by filtration, washed with methanol and dried in the vacuum oven to constant weight. The monomer conversion was calculated from the polymer weight and the initial amount of the monomer. The procedure was the same for the solution polymerization tests, except for the fact that a solvent, toluene, was used. Toluene was added to the Schlenk tube at room temperature under argon together with other components of the reaction mixture.
Polymerization of L,D-lactide
The bulk polymerization of L,D-lactide was carried out in a Schlenk tube where the complex and the appropriate amount (resulting from the assumed monomer/transition metal molar ratio) of L,D-lactide were charged under argon. Then, the tube was placed on the oil bath which had been pre-heated to 150 °C and the mixture was stirred at that temperature for a specified amount of time. After the reaction was quenched with the use of methanol, the reaction mixture was dissolved in a minimum amount of CH2Cl2 and transferred to cold methanol. Some part of the solvents was removed under vacuum and the polymer was separated by filtration, washed with methanol and dried in the vacuum oven to constant weight. The monomer conversion was calculated from the weight of the polymer and the initial amount of the monomer.
Results and discussion
Synthesis of complexes
Ligand precursors LH2 (Scheme 1) was prepared by reaction of 3,5-di-tert-butyl-2-hydroxybenzaldehyde with the appropriate amine (2-aminobenzyl alcohol) in refluxing ethanol according to the methods described in the literature [50, 51]. The compound was isolated in 69% yields by flash chromatography and analyzed by FTIR, 1H and 13C NMR spectroscopy to confirm its structure. In the 1H NMR spectrum of LH2 (Fig. 1) characteristic signals due to proton of imine and hydroxyl group were displayed as singlets at 8.62 ppm and 13.39 ppm, respectively. Signal of benzylic protons was also singlet and was located at 4.90 ppm.

Synthesis of ligand and corresponding complexes

1H NMR spectra of LH2 and corresponding titanium (LTi) and vanadium (LV) complexes (* residual deuterated solvent peak, ^ toluene)
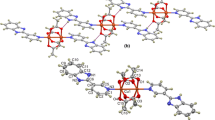
The group 4 and 5 transition metal complexes were prepared via the direct reaction of appropriate metal isopropoxide (Ti(OiPr)4, VO(OiPr)3) with the synthesized tridentate Schiff base ligand precursor in the 1:1 molar ratio (Scheme 1). The reactions of both metal alkoxides with LH2 were carried out in toluene at room temperature, as it is typically done, and proceeded readily with high yields (68.9–87.4%). The NMR spectra (Fig. 1 and Figure S2) of the products confirmed the formation of complexes. No spectrum of any complex contained the signal for the proton in the –OH group which could be observed in the spectrum for the ligand at 13.39 ppm. It can be also seen that spectrum of LTi showed very complex signals. Primarily, the chemical shift for the imine proton was registered at 8.47 ppm, 8.43 ppm and at 8.42 ppm. In addition, the multiple signals for tBu and ispropoxy groups and other protons suggested the formation of the isomers. Crystallization from toluene allowed to obtained single crystal of LTi and X-ray studies revealed the dimeric structure of this isomer (Fig. 2). The analysis showed also that in the asymmetric part of the unit cell the main product and a disordered toluene molecule as a solvent are present. In addition, two iPr groups are also disordered. In both cases, the disorder is typical. The disordered fragments were removed from the drawings to keep the image more clear and to better show the core structure of the resulting complex with octahedral (NOC-5) geometry of the Ti metal centre. The central core is fairly regular for various compounds with the average Ti − Ti distance of 3.3050(8) Å, from a small range of distances 2.74 − 3.296 Å [52]. The Ti − OR distances fall within a relatively small range (1.8002(16) – 2.0926(16) Å) and they are consistent with the literature reported figures [53]. As stated previously, the central Ti2O2 core is fairly regular with the average Ti − O − Ti angles of 108.40(7)° and O − Ti − O angles of 71.60(7)° – these are consistent with the literature reported values. The remainder of the angles around the Ti atom is best described as a similar distorted OC-6 geometry [53]. The other parameters for the crystal structure are given in Table S2. In case of LV 1H NMR spectrum showed one main product with small admixture of a few percent of an isomer. The signal of the imine proton appeared upfield in respect to the appropriate signal for the ligand, e.g. as a singlet at 8.53 ppm. The spectrum showed also two singlets for tert-butyl groups (at δ = 1.35 and 1.49 ppm), two doublets at 5.44 and 5.67 ppm assigned to the methylene protons of the OCH2Ar group, and the signals of protons in O–CH(CH3)2 group appeared in the spectra for the complex at 5.79–5.86 ppm. The signals for the aromatic protons were also shifted upon complexation, for example doublets at 7.24 and 7.48 ppm in the spectrum of the ligand characteristic for protons of substituted by tBu aromatic ring moved to 7.32 and 7.64 ppm. Unfortunately, we did not obtained crystals suitable for X-ray analysis. The evidence for the formation of the complexes rises from the analysis of the FTIR spectra of LH2 and corresponding complexes as well (Figure S3). The spectrum of the ligand shows inter alia a broad absorption band at 3311 cm−1 which can be assigned to the hydrogen bond ν(O–H) and a medium intensity band at 1616 cm−1 due to the azomethine ν(C = N) linkage. The first band disappears on the complexation and the other shows a shift to lower wave numbers by 5–4 cm−1 to 1611 and 1612 for LTi and LV, respectively, suggesting the involvement of the azomethine nitrogen atom in chelation to the metal ion.

(a) Arrangement of the complex molecule in the unit cell. Disordered toluene molecule as solvent and disorder iPr groups omitted for clarity. (b) Molecular structure of LTi in the asymmetric part of unit cell. Displacement ellipsoids are drawn at the 50% probability level. Disordered toluene molecule as solvent and disorder iPr groups are omitted for clarity
Polymerization of ε-caprolactone
The ring opening polymerizations of ε-caprolactone with the use of synthesized complexes were carried out under various reaction conditions in bulk. Some polymerization tests were also performed in toluene for reference. The results are summarized in Table 1. The initial tests to verify the effect of various process parameters on CL polymerization were made with LTi. The obtained results showed that the conversion of CL to PCL was significantly affected by the molar ratio of the monomer to the LTi complex. The conversion sharply decreased with the increasing CL/Ti from 100 to 250 and to 600 in the polymerization processes at 100 °C (runs 1–3) as well as with the increase of CL/Ti from 600 to 1600 in the polymerization tests at 120 °C (runs 4 and 5). The influence of the solvent on the polymerization results was verified in the reactions which were carried out for 50 min at CL/Ti = 600 and at 100 °C (runs 6 and 7). The complex performed better in the absence of toluene and it gave the monomer conversion 38.7% and Mn = 10.4·103 g/mol (Mw/Mn = 1.6). For polymerization in solution conversion was 25% and Mn = 8.1·103 g/mol (Mw/Mn = 1.5). The effect of temperature on the polymerization reaction in the presence of LTi was investigated at CL/Ti (molar ratio) = 600:1 (Table 1, runs 4, 6, 8 and 9). The increase of temperature was found to improve the reaction efficiency. After 50 min, no poly(ε-caprolactone) was formed in practice at 25 °C. The monomer conversion increased to 15.8% and to 38.7% at 70 °C and at 100 °C, respectively. However, the highest conversion (86.7%) and thereby the polymerization rate was obtained at 120 °C after just 19 min. Comparison of obtained results with those reported in literature for ε-caprolactone polymerization in the presence of other titanium complexes with tridentate ligands (Table S4) showed good performance of LTi.
The molar mass value of the polymers obtained with LTi was also dependent on the reaction temperature. It increased from 6.6·103 to 18.7·103 g/mol as the reaction temperature increased from 70 to 120 °C. Typical SEC chromatograms are shown in Fig. 3. The experimental values of Mn were compared with the theoretical ones calculated from the equation: molar mass of the monomer × ([monomer]0/2[Ti]0) × conversion yield + 60.10 [10, 28]. The theoretical number-average molar mass (Mn(theo)) of the polymers synthesised in bulk at 70 °C and 100 °C (5.5·103 g/mol for run 9 and 13.3·103 g/mol for run 6) as well as Mn(theo) of PCL synthesized in solution (8.6·103 g/mol, run 7) well correlated with the experimental data included in Table 1. Moreover, the dispersity of PCLs produced by LTi is quite low (Mw/Mn = 1.4–1.7). The only exception made to this rule is PCL synthesized at 120 °C in bulk (run 4). Its dispersity is much wider (Mw/Mn = 2.5) and its molar mass differs significantly from the expected value (Mn(theo) for run 4 = 29.7·103 g/mol). This deviation may result from both the increased viscosity due to high conversion and side reactions at elevated temperatures. It is known that a higher temperature favors the occurrence of side reactions which are indeed typical for ROP of cyclic esters, such as inter- and intramolecular reactions, namely reshuffling and backbiting reactions, respectively [54].

SEC chromatograms of PCLs produced by LTi (sample designation as in Table 1)
The results for the ROP of ε-caprolactone presented in Table 1 showed that the change of the complex from LTi to LV leads to a decline in the monomer conversion. Despite a few times longer polymerization time (60 min), the conversion value for CL at 120 °C and at CL/M = 600 equals to 3.8% in presence of LV, while the titanium catalyst gave 86.7% after 19 min. However, much longer reaction times increased the monomer conversion: up to 42.0% after 300 min (Fig. 4, runs 11–14). For the same reaction time but at a higher CL/V molar ratio (1200), the monomer conversion was equal to 19.1% (run 15). Selected data on ε-caprolactone polymerization in the presence of different vanadium catalysts reported in literature were compared in Table S3. As can be seen, it is difficult to directly compare the results with those described in literature for ROP of CL with the use of other vanadium compounds because of much different reaction conditions. However, the vanadium catalysts investigated so far displayed rather poor or moderate activity in that process [9, 41,42,43, 55]. Taking into account the high CL/V molar ratio and much shorter polymerization time (5 h) than usually applied for the vanadium catalysts (24 h or more), the obtained results confirmed good efficiency of LV. It should be noticed that LV remains active also at 100 °C but its time-conversion curves (Fig. 4) showed that the reaction rate decreased at the declining temperatures. However, monomer conversion increases progressively with the increasing polymerization time (Fig. 4, runs 16–18) to 19.9% after 360 min. The monomer conversion increases also with the decrease of the CL/V ratio to 250 and to 100 (runs 19 and 20) and it reaches 13.5% at CL/V = 100 after two hours. The resulting PCLs have low molar masses (4·103—7.4·103 g/mol) and that value, as can be observed from Fig. 5 and Table 1, increases with time irrespective of the reaction temperature. Moreover, Mn values determined by SEC are in good agreement (difference between 0 and 11%) with those calculated from NMR spectra (Table 1). The molar mass distribution for PCLs is in the range 1.4–1.6 and it is monomodal (Fig. 6). The experimental values of Mn for PCLs obtained at 120 °C and CL/V = 600 after various reaction times were compared with the theoretical values calculated with the assumption that one isopropoxide group initiates the polymerization process. The Mn(theo) values equal to 21.3·103 g/mol (run 12), 27.3·103 g/mol (run 13) and 28.8·103 g/mol (run 14) were significantly higher than the experimental ones. That suggests that the ROP of CL with the vanadium complex at 120 °C is not well controlled.

Effect of reaction time on the conversion of ε-caprolactone in presence of LV at the molar ratio of CL/V equal to 600:1 in the bulk at 100 °C (■) and 120 °C (●)

Plot of Mn of PCL as a function of monomer conversion in polymerization catalyzed by LV (at 100 °C (■) run 17, 18 and at 120 °C (●) run 12–14)

Evolution of the SEC chromatograms of PCLs produced by LV at 120 °C and at 100 °C with the reaction time (runs 12–14 and 18 from Table 1)
Selected poly(ε-caprolactone)s obtained in the presence of LTi and LV were analysed by 1H and 13C NMR spectroscopy. The recorded spectra (the exemplary ones are presented in Fig. 7) looked very similar regardless of the type of the complex which was used in the synthesis. The 13C NMR spectra showed the signals of carbons 4–8 of the repeating units of poly(ε-caprolactone) at 24.71 (C6), 25.67 (C5), 28.49 (C7), 34.26 (C4), 64.27 (C8) and 173.65 (C3). The signals were assigned on the basis of the previous work [33, 56]. The 1H NMR spectra showed main peaks at 1.35, 1.64, 2.30 and 4.05 ppm which were due to methylene protons at carbons 4–8 of the repeating units of PCL and weak signals corresponding to end-groups. Obviously, the triplet appeared at 3.64 ppm is due to methylene protons of the -CH2-OH. Such terminal group is formed when the reaction is terminated by hydrolysis. The presence of signals for the protons of the iso-propyl group at 1.22 ppm (doublet, (CH3)2CHO-) and 5.00 ppm (multiplet, (CH3)2CHO-) indicate that propagation proceeded by coordination of the monomer to the active species and then its insertion into metal–oxygen bond. This can imply that the polymerization reaction followed the coordination-insertion mechanism, alike for other transition metal complexes having the alkoxide groups [9, 33].

1H (a) and 13C (b) NMR spectrum of a typical poly(ε-caprolactone) sample (PCL obtained in run 14 in the presence of LV)
The thermal properties of the produced poly(ε-caprolactone)s were investigated by means of the DSC analysis. In each case the DSC thermogram (Fig. 8 displayed the exemplary ones) revealed the presence of a single narrow endothermic melting peak of PCL with the minimum at 52.9 °C – 56.8 °C and crystallinity in the range 49.0–60.2%.

DSC thermograms of poly(ε-caprolactone)s obtained with LV (run 13 and 17) and with LTi (run 4 and 6)
The better performing in polymerization of CL titanium complex was also tested for its ability to polymerize L,D-lactide. The bulk polymerization test was carried out at 150 °C, at LA/Ti = 154 for 115 min. Although the polymerization conditions were not optimized, that complex initiated the polymerization process and the monomer conversion obtained from the weight of the polymer and the initial amount of the monomer was around 22%. The formation of PLA was confirmed by the 13C NMR (Figure S1) analysis. The obtained amorphous polymer had the glass transition temperature equal to 44.3 °C. It seems that the presented complex after process optimization can be efficiently used for polymerization of various lactones but that assumption has to be verified in the future research.
Conclusions
Titanium and vanadium alkoxides supported by phenoxy-imine ligand with the additional O-donor group were synthesised and used for ROP of ε-caprolactone. Moderate to very good conversion figures at 120 °C were achieved in the ROP of CL with the use of the synthesized complexes, with the molar mass (Mn) and dispersity of PCLs in the range of ~ 4·103 to 18.7·103 g/mol and 1.3–1.7, respectively. The titanium complex offered the catalytic activity which was significantly higher than that for the vanadium one. For example, activity of LTi at CL/Ti = 600 and 120 °C was equal 187 gpol mmolTi−1 h−1 (run 4) while activity of vanadium complex at the same reaction conditions was equal 7 gpol mmolV−1 h−1 (run 12). It was also shown that the titanium complex can be used in polymerization of lactide.
Data availability
The data used to support the findings of this study are included within the article.
References
Gupta B (2012) Geeta, Ray AR. J App Polym Sci 123:1944–1950
Labet M, Thielemans W (2009) Chem Soc Rev 38:3484–3504
Woodruff MA, Hutmacher DW (2010) Prog Polym Sci 35:1217–1256
Guarino V, Gentile G, Sorrentino L, Ambrosio L (2017) Encyclopedia of Polymer Science and Technology. John Wiley & Sons
Mohamed RM, Yusoh K (2015) Adv Mat Res 1134:249–255
Dash TK, Konkimalla VB (2012) J Control Release 158:15–33
Okada M (2002) Prog Polym Sci 27:87–133
Li J, Deng Y, Jie S, Li BG (2015) J Organometal Chem 797:76–82
Arbaoui A, Redshaw C (2010) Polym Chem 1:801–826
Liu H, Khononov M, Fridman N, Tamm M, Eisen MS (2019) Inorg Chem 58:13426–13439
Zhao W, Wang Q, Cui Y, He J, Zhang Y (2019) Dalton Trans 48:7167–7178
Huang YT, Wang WC, Hsu CP, Lu WY, Chuang WJ, Chiang MY, Lai YC, Chen HY (2016) Polym Chem 7:4367–4377
Xi Y, Rajendran NM, Zhang W, Sun Y, Lei M, Sun WH (2016) Chemistry Select 1:5660–5665
Huang HC, Wang B, Chen XL, Pan L, Li YS (2019) J Polym Sci Part A: Polym Chem 57:973–981
Liu S, Zhang J, Zuo W, Zhang W, Sun WH, Ye H, Li Z (2017) Polymers 9:83
Pei H, Yang H, Lu N, Liu W, Li Y (2017) Z Anorg Allg Chem 643:511–515
Kiriratnikom J, Chotchatchawankul S, Haesuwannakij S, Kiatisevi S, Phomphrai K (2018) New J Chem 42:8374–8383
Lee CL, Lin YF, Jiang MT, Lu WY, Vandavasi JK, Wang LF, Lai YC, Chiang MY, Chen HY (2017) Organometallics 36:1936–1945
Zaitsev KV, Cherepakhin VS, Zherebker A, Kononikhin A, Nikolaev E, Churakov AV (2018) J Organometal Chem 875:11–23
Liu ZT, Li CY, Chen JD, Liu WL, Tsai CY, Ko BT (2017) J Mol Struct 1134:395–403
Chen XL, Wang B, Pan L, Li YS (2019) App Organometal Chem 33:e4770
Żółtowska K, Sobczak M, Olędzka E (2015) Molecules 20:2816–2827
Lai FJ, Chiu LL, Lee CL, Lu WY, Lai YC, Ding S, Chen HY, Wu KH (2019) Polymer 182:121812
Cota I (2017) Phys Sci Rev 2:20160129
Lyubov DM, Tolpygin AO, Trifonov AA (2019) Coord Chem Rev 392:83–145
Takashima Y, Nakayama Y, Hirao T, Yasuda H, Harada A (2004) J Organometal Chem 689:612–619
Chmura AJ, Davidson MG, Jones MD, Lunn MD, Mahon MF (2006) Dalton Trans 887–889
Jiang MT, Kosuru SR, Lee YH, Lu WY, Vandavasi JK, Lai YC, Chiang MY, Chen HY (2018) eXPRESS Polym Lett 12: 126–135
Chmura AJ, Davidson MG, Jones MD, Lunn MD, Mahon MF, Johnson AF, Khunkamchoo P, Roberts SL, Wong SSF (2006) Macromolecules 39:7250–7257
Kuchuk EA, Mankaev BN, Zaitsev KV, Oprunenko YF, Churakov AV, Zaitseva GS, Karlov SS (2016) Inorg Chem Commun 67:1–5
Durr CB, Williams CK (2018) Inorg Chem 57:14240–14248
Schwarz AD, Thompson AL, Mountford P (2009) Inorg Chem 48:10442–10454
Ou HW, Lu WY, Vandavasi JK, Lin YF, Chen HY, Lin CC (2018) Polymer 140:315–325
Dakshinamoorthy D, Peruch F (2011) J Polym Sci Part A: Polym Chem 49:5176–5185
Chakraborty D, Mandal D, Ramkumar V, Subramanian V, Sundar JV (2015) Polymer 56:157–170
Piskun YI, Vasilenko IV, Kostjuk SV, Zaitsev KV, Zaitseva GS, Karlov SS (2010) J Polym Sci: Part A: Polym Chem 48:1230–1240
Piskun YI, Vasilenko IV, Zaitsev KV, Karlov SS, Zaitseva GS, Gaponik LV, Kostjuk SV (2015) Russ Chem Bull 64:181–188
Saha TK, Ramkumar V, Chakraborty D (2011) Inorg Chem 50:2720–2722
Ou HW, Chen HY, Tseng HC, Hsiao MW, Chang YL, Jheng NY, Lai YC, Shih TY, Lin YT, Chen HY (2014) J Mol Cat A: Chem 394:97–104
Gupta KC, Sutar AK (2008) Coord Chem Rev 252:1420–1450
Ge F, Dan Y, Al-Khafaji Y, Prior TJ, Jiang L, Elsegood MRJ, Redshaw C (2016) RSC Adv 6:4792–4802
Clowes L, Walton M, Redshaw C, Chao Y, Walton A, Elo P, Sumerin V, Hughes DL (2013) Catal Sci Technol 3:152–160
Arbaoui A, Redshaw C, Homden DM, Wright JA, Elsegood MRJ (2009) Dalton Trans 8911–8922
Clowes L, Redshaw C, Hughes DL (2011) Inorg Chem 50:7838–7845
Save M, Schappacher M, Soum A (2002) Macromol Chem Phys 203:889–899
(a) CrysAlis CCD; Oxford Diffraction Ltd: Abingdon, England, (2002) (b) CrysAlis RED. Abingdon, England, Oxford Diffraction Ltd, p 2002
Sheldrick GM (2008) Acta Crystallogr. Sect A 64:112–122
Sheldrick GM (2015) Acta Crystallogr. Sect C 71:38
Macrae CF, Bruno IJ, Chisholm JA, Edgington PR, McCabe P, Pidcock E, Rodriguez-Monge L, Taylor R, van de Streek J, Wood PA (2008) J Appl Crystallogr 41:466–470
Sierra MA, Gómez-Gallego M, Alcázar R, Lucena JJ, Yuntab F, García-Marco S (2004) Dalton Trans 3741–3747
Karakas A, Elmali A, Ünver H, Svoboda I (2004) J Mol Struct 702:103–110
Groom CR, Bruno IJ, Lightfoot MP, Ward SC (2016) Acta Crystallogr, Sect. B: Struct Sci. Cryst Eng Mater B72:171–179
Boyle TJ, Guerrero FJ, Alam TM, Dunnigan KA, Sears JM, Wheeler DR (2020) Inorg Chem 59:880–890
Schmid M, Guillaume SM, Roesky PW (2014) Organometallics 33:5392–5401
Ma J, Zhao KQ, Walton M, Wright JA, Hughes DL, Elsegood MRJ, Michiue K, Sun X, Redshaw C (2014) Dalton Trans 43:16698–16706
Cayuela J, Bounor-Legaré V, Cassagnau P, Michel A (2006) Macromolecules 39:1338–1346
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the is no conflicts of interest regarding publication of this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Białek, M., Fryga, J., Spaleniak, G. et al. Ring opening polymerization of ε-caprolactone initiated by titanium and vanadium complexes of ONO-type schiff base ligand. J Polym Res 28, 79 (2021). https://doi.org/10.1007/s10965-021-02419-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-021-02419-y