
Abstract
We have investigated the performance of the dispersion-corrected density functionals (BLYP-D, BP86-D and PBE-D) and the widely used B3LYP functional for describing the hydrogen bonds and the stacking interactions in DNA base dimers. For the gas-phase situation, the bonding energies have been compared to the best ab initio results available in the literature. All dispersion-corrected functionals reproduce well the ab initio results, whereas B3LYP fails completely for the stacked systems. The use of the proper functional leads us to find minima for the adenine quartets, which are energetically and structurally very different from the C4h structures, and might explain why adenine has to be sandwiched between guanine quartets to form planar adenine quartets.
Similar content being viewed by others
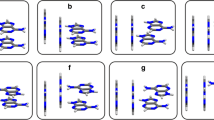
1 Introduction
Besides the well-known Watson–Crick pairing of nucleic acid bases in the helical DNA molecule, these bases are known in telomeres to form cyclic hydrogen-bonded quartets [1–11] (see Scheme 1), which, in turn, are able to stack together through π-stacking interactions. These quartet structures of DNA bases are increasingly recognized for their biological importance [1–11].

Guanine and adenine quartets
By far the longest-known is the guanine quartet (G4), which has an exceptional stability due to its capacity to form two hydrogen bonds between neighboring guanines, i.e., the N1–H···O6 and N2–H···N7 hydrogen bonds (see Scheme 1). Furthermore, the stabilization of guanine quartets can be enhanced by cations [2]. This high stability of G4 facilitates through stacking the formation of other quartets, for instance of A4 [4–7]. However, the experimental evidence for the existence of adenine quartets is rare. To our knowledge, adenine quartets have only been observed experimentally sandwiched between two guanine quartets in [4–7]. The adenine quartets are formed with one hydrogen bond between adjacent bases (see Scheme 1). The three quartets have as proton-donor the N6–H bond of the amino-group and as proton-acceptor the N1 atom (A4-N1) [4, 5], the N3 atom (A4-N3) [6], and the N7 atom (A4-N7) [7], respectively. The guanine quartet, as well as the three adenine quartets, has been the subject of computational studies [12–19].
In this work, we analyze the stability of these four quartets in the gas phase and in aqueous solution using dispersion-corrected density functional theory (DFT-D). First, we explore the performance of different DFT-D variants for the hydrogen-bonded (see Scheme 2) and stacked DNA base pairs (see Fig. 1, later on) by comparing them with high-level ab initio results [20–22]. In our previous work, we showed that the hydrogen bonds between adjacent bases from opposite strands in DNA are described adequately by a number of GGA, meta-GGA and hybrid DFT approaches (for instance BP86) [23, 24]. However, most of the current DFT approaches, ranging from GGA via meta-GGA to hybrid DFT, fail in describing π-stacking interactions between two bases within each of the two DNA strands [25, 26].

Watson–Crick base pairs

Stacked AT and GC dimers in the gas phase shown from two side views (BLYP-D/TZ2P)
After having established the proper dispersion-corrected functional, we investigate for the four quartets three different configurations which are of C4h, C4 and S4 point group symmetry and which all have the cyclic hydrogen bonds as shown in Scheme 1. The adenine quartets have been observed experimentally in a planar configuration [4–7], and we examine whether these quartets remain in a planar configuration when the stabilization by the guanine quartets is not present. Finally, we test the applicability of the widely used B3LYP functional for these quartets in the different symmetries and we show that the neglect of the dispersion correction will even lead to erroneous conclusions.
2 Computational methods
All calculations were performed using the Amsterdam density functional (ADF) program developed by Baerends et al. [27–38], and the QUantum-regions Interconnected by Local Descriptions (QUILD) program by Swart and Bickelhaupt [39, 40]. The QUILD program is a wrapper around ADF (and other programs) and is used for its superior geometry optimizer which is based on adapted delocalized coordinates [39]. The numerical integration was performed using the procedure developed by te Velde et al. [34, 35].
The MOs were expanded in a large uncontracted set of Slater type orbitals (STOs) containing diffuse functions: TZ2P (no Gaussian functions are involved) [36]. The basis set is of triple-ζ quality for all atoms and has been augmented with two sets of polarization functions, i.e., 3d and 4f on C, N, O and 2p, 3d on H. The 1s core shells of carbon, nitrogen and oxygen were treated by the frozen-core approximation [30]. An auxiliary set of s, p, d, f and g STOs was used to fit the molecular density and to represent the Coulomb and exchange potentials accurately in each self-consistent field cycle [37].
Calculations were done using dispersion-corrected DFT-D as developed by Grimme [41–47] for a correct treatment of the stacking interactions between the DNA bases. The density functionals are augmented with an empirical correction for long-range dispersion effects, described by a sum of damped interatomic potentials of the form C6R−6 added to the usual DFT energy [41–47]. Equilibrium structures were optimized using analytical gradient techniques [38]. Geometries and energies were calculated with the generalized gradient approximation (GGA) at BLYP [48, 49], BP86 [48, 50] and PBE [51] as well as with the dispersion-corrected variants BLYP-D, BP86-D and PBE-D. Furthermore, the hybrid functional M06-2X [52] has been used for the computation of geometries and energies of AT and GC (both hydrogen-bonded and stacked dimers) and for the systems with stacking interactions involved. For the quartets the B3LYP [49, 53, 54] has been used to compute energies in a single-point fashion using the BLYP-D geometries.
At the BLYP-D level of theory, all energy minima of hydrogen-bonded AT and GC pairs, stacked AT and GC dimers, and the global energy minima of DNA-base quartets have been verified in the gas phase and in water to be equilibrium structures through vibrational analysis [55–57]. The lowest energy minima were found to have zero imaginary frequencies (see also Electronic Supplementary Material). For the dispersion-corrected functionals the basis set superposition error (BSSE) on the bond energy was not calculated because the dispersion correction [42] has been developed such that the small BSSE effects are absorbed into the empirical potential. For the bond energy calculated with the uncorrected functionals the BSSE has been computed and corrected through the counterpoise method [58], using the individual DNA bases as fragments.
Solvent effects in water have been estimated using the conductor-like screening model (COSMO) [59, 60], as implemented in the ADF program [61]. For settings see [62]. The continuum solvent model performs adequately for the determination of geometries as has been done in this work. However, for the calculation of spectra of molecules in a solvent, it is essential to take into account the first shell of solvent molecules explicitly [63–69]. For instance, Nicu et al. [63] showed that the VCD spectrum of 2-benzoic acid is influenced by the hydrogen bonding with the solvent due to the donor–acceptor interaction (which of course is not taken into account by continuum solvent models). According to the work by Riley et al. [70] the dispersion correction does not need to be modified for the solvated systems.
3 Results and discussion
3.1 AT and GC pairs
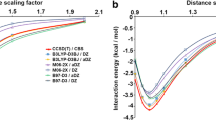
The hydrogen-bond distances and energies of the Watson–Crick base pairs AT and GC in the gas-phase and in aqueous solution are given in Tables 1 and 2, respectively, together with the best ab initio estimate by Sponer et al. [20]. The base pairs have been optimized in Cs symmetry and the bases in C1 in accordance with our previous work [23, 24]. The hydrogen bond energy for AT and GC is defined as the difference in energy between the optimized pair and the fully optimized bases. From Tables 1 and 2, we see that the hydrogen bond energies for AT and GC obtained with the dispersion-uncorrected density functionals, including the M06-2X functional, are all a few kilocalorie per mole more weakly bound than the CCSD(T) reference value with a clear underestimation of the hydrogen-bond energies by the B3LYP functional. The hydrogen-bond energies acquired with the dispersion-corrected density functionals are all slightly more strongly bound than the CCSD(T) reference value. Although the BP86-D and the PBE-D results already agree well with the ab initio results, the best agreement with CCSD(T) is obtained with BLYP-D. At BLYP-D/TZ2P, the hydrogen-bond energy for the Watson–Crick AT and GC pairs amounts to −16.7 and −30.1 kcal/mol, which has to be compared with −15.4 and −28.8 kcal/mol, respectively, at CCSD(T)/aug-cc-pVQZ//RI-MP2/cc-pVTZ [20–22]. This close agreement between dispersion-corrected functionals and ab intio results for the hydrogen-bonded and stacked AT and GC pair was also found by Grimme and co-workers [43] with Gaussian-type triple and quadruple-zeta basis sets. Furthermore, the inclusion of water as a solvent has been investigated. The hydrogen bonds between the AT and GC in water are half as strong as the same hydrogen bonds in the gas phase.
The π-stacking interaction in the AT and GC dimers in the gas phase and in aqueous solution are given in Table 3, together with the best ab initio estimate by Sponer et al. [20]. For the stacked AT and GC pairs, the dispersion corrected functionals are in close agreement with the ab initio results. The M06-2X energies are about 2 kcal/mol too weakly bound, which has also been found by Kabelac et al. [71, 72] for the M05-2X functional. The BLYP-D energies of −11.7 and −16.9 kcal/mol, respectively, are in excellent agreement with the CCSD(T)/aug-cc-pVQZ//RI-MP2/TZVPP ones of −11.6 and −16.9 kcal/mol, respectively [20–22].
The B3LYP functional appears to be completely inadequate for describing stacked DNA bases, similar to what we have found previously for regular GGA functionals such as BP86 [25]. This follows from numerical experiments in which we evaluate the stacking energies in the gas phase and in water with B3LYP/TZ2P at the BLYP-D/TZ2P equilibrium structures, i.e., B3LYP/TZ2P//BLYP-D/TZ2P. In the gas phase, this yields for the stacked AT a slightly repulsive result of 2.1 kcal/mol. For GC the stacking energy in the gas phase amounts to −7.5 kcal/mol. The same numerical experiment has been done for PBE, which gives the AT pair bonded by only 1.4 kcal/mol and the GC pair by 10.1 kcal/mol at the PBE/TZ2P//BLYP-D/TZ2P level. Note that the reason for the attractive B3LYP and PBE energies of stacked GC is the presence of two hydrogen-bonding like interactions in the stacked GC system in which G and C are, in fact, not exactly parallel but under a slight angle. This is because of the aforementioned “partial” hydrogen bonds which B3LYP or PBE are able to describe. Stacked GC differs in this respect from stacked AT which is bound by a more pure π-stacking interaction which B3LYP or PBE is not able to describe properly (see Fig. 1). The stacked GC pair is therefore not a good benchmark to evaluate the adequacy of functionals for stacking interactions. However, starting from a stacked AT or GC pair the optimization with the B3LYP or PBE functional leads eventually to a hydrogen bonded base pair because of the erroneous description of the stacking interactions by these functionals. In water, both the AT and GC stacks would, according to B3LYP, not exist, which is evidently incorrect.
3.2 Adenine and guanine quartets
The results of our BLYP-D/TZ2P study on the formation of the G4, A4-N1, A4-N3 and A4-N7 quartets in the point group symmetries C4h, C4 and S4 in the gas phase and in water are summarized in Table 4 (energies), Figs 2, 3, 4, 5 and Tables S1–S4 of the ESM (geometries). The geometries are in both phases almost similar, except for the A4-N3 minima in the S4 symmetry. For the C4h symmetry the hydrogen bond lengths are given for the gas phase and the condensed phase in Fig. 2. In the C4 symmetry we find for all quartets bowl-shaped structures (see Fig. 3). The S4 structures in water are presented in Fig. 4. For G4 and A4-N3 quartets the systems are slightly distorted from planarity and for A4-N1 and A4-N7 we find stacked dimers with cyclic hydrogen bonds in between (for A4-N1 with four N6(H)···N1 and for A4-N7 with four N6(H)···N7 hydrogen bonds as presented in Scheme 1). The number of imaginary frequencies from the vibrational analysis of the structures in different symmetries can be found in Table S6 of the ESM. The hydrogen bond energies for B3LYP have been calculated with the BLYP-D geometries to give a qualitative impression of how B3LYP is unable to reproduce energies for stacked complexes (see Table 4).

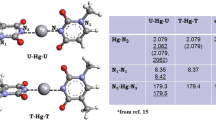
The C4h structures in water of G4, A4-N1, A4-N3 and A4-N7 at the BLYP-D/TZ2P level of theory. Hydrogen-bond distances (Å) are given for aqueous solution (gas-phase values in parentheses)

Side view and top view of the C4 structures in water of A4-N1, A4-N3 and A4-N7 at the BLYP-D/TZ2P level of theory

Structures of the S4 global minima in water of G4, A4-N1, A4-N3 and A4-N7 at the BLYP-D/TZ2P level of theory (for G4 and A4-N3 a side and a top view are shown)

Side view of the local S4 minimum in the gas phase of A4-N1 at the BLYP-D/TZ2P level of theory
In accordance with previous work of Meyer et al. [18] at the B3LYP level, we find in the gas phase for the G4 quartet that the difference between the C4h and the S4 structures is small, only 0.6 kcal/mol (see Table 4; Fig. 4). Also for the A4-N3 quartet, we find geometrically equivalent structures as in previous work [17] at the B3LYP level. However, we find that all structures are energetically very similar (within 0.4 kcal/mol equal), whereas Meyer et al. [17] found that at the B3LYP level the S4 structure is 8.6 kcal/mol higher in energy than the C4h structure.
For the A4-N1 and the A4-N7 quartets, we find that the S4 structure is more than 16 and 12 kcal/mol, respectively, lower in energy than the other symmetric structures. This is in sharp contrast with previous work [18] at the B3LYP level, where the S4 structure of A4-N1 is only a few kilocalorie per mole lower in energy than the C4 and C4h structures, and for the A4-N7 quartet all symmetric structures are within 0.5 kcal/mol energetically equal. The difference can be ascribed to the difference in the geometries. Our S4 minima are stacked dimers with hydrogen bonds in between, whereas the B3LYP minima [18] look more like a distorted C4h structure. For the A4-N1 quartet we were able to find a local minimum in the S4 symmetry at the BLYP-D/TZ2P level (see Fig. 5), which is very similar to the B3LYP structure. The bond energy of the local S4 minimum amounts only to −31.2 kcal/mol, while the bond energy of the global S4 minimum is −46.0 kcal/mol. To analyze if the B3LYP functional could reproduce this result, we calculated the B3LYP/TZ2P bonding energies with our BLYP-D/TZ2P structures: the bond energy of the stacked dimers with hydrogen bonds in between (Fig. 4) amounts then to −9.7 kcal/mol and of the “distorted C4h” structure (Fig. 5) to −14.4 kcal/mol. Thus, in addition to a general underestimation of the stability of our stacked model systems, the B3LYP functional also yields the wrong energetic ordering of the minima (see also Table 4). This demonstrates once more how the B3LYP functional leads to qualitatively wrong chemical conclusions.
3.3 To stack or not to stack
In aqueous solution, our results show again that for the G4 and the A4-N3 all three symmetries are very close in energy and that it costs only 0.2 kcal/mol in both cases to go from the S4 geometry (as presented in Fig. 4) to the flat C4h symmetric geometry. Such planarization is necessary in order to form stacked systems.
The bonding energy (−33.6 kcal/mol) of the G4 quartet in the C4h symmetry is twice as large as the bonding energy (−16.8 kcal/mol) of the A4-N3 quartet and therefore G4 is, as expected, more stable. The A4-N1 and the A4-N7 quartets bind in the S4 geometry with −33.2 and −27.2 kcal/mol, respectively, which is of the same magnitude as the bonding energy of G4. To go from the nonplanar stacked system in the S4 geometry (see Fig. 4) to the planar C4h geometry, which is needed to form stacks of quartets, it costs 22.3 kcal/mol for the A4-N1 quartet and 11.0 kcal/mol for the A4-N7 in aqueous solution.
The relatively low stability of planar A4-N1 and A4-N7 quartets and their strong tendency to form S4-symmetric nonplanar arrangements might be the decisive factor behind the fact that these A4 quartets are not so often observed experimentally. This also explains why these quartets are found sandwiched between two G4 quartets, which supplies the required stabilization of the planar configuration through stacking interactions.
4 Conclusions
We have shown that the dispersion-corrected density functionals, especially BLYP-D, are very well suited for describing both hydrogen bonded AT and GC pairs and stacked AT and GC dimers, with bond energies and distances that are in excellent agreement with the best ab initio CCSD(T) benchmark data in the literature.
The B3LYP functional fails completely to describe the stacked AT and GC dimers. Furthermore, B3LYP fails to reproduce the strong tendency of A4 quartets to adopt nonplanar structures. Our findings imply that B3LYP should not be used for DNA systems that involve π-stacking interactions.
Our BLYP-D results show that in water G4 is the most strongly bound quartet and easily adopts a planar geometry. The planar geometries of all A4 quartets studied are two to three times less stable than planar G4. In addition, by far the most stable A4 structure is nonplanar. In other words, A4 has, unlike G4, a strong tendency to adopt a geometry that is unsuitable for stacking. This explains the fact that A4 quartets have been experimentally observed in stacks only in between G4 quartets: they are less stable and nonplanar and therefore need the stabilization of planar G4 in order to form larger stacks.
References
Neidle S, Balasubramanian S (eds) (2006) Quadruplex Nucleic Acids. RSC Publishing, Cambridge
Davis JT (2004) Angew Chem Int Ed 43:668
Gros J, Rosu F, Amrane S, De Cian A, Gabelica V, Lacroix L, Mergny JL (2007) Nucleic Acids Res 35:3064
Searle MS, Williams HEL, Gallagher CT, Grant RJ, Stevens MFG (2004) Org Biomol Chem 2:810
Patel PK, Koti ASR, Hosur RV (1999) Nucleic Acid Res 27:3836
Pan B, Xiong Y, Shi K, Sundaralingam M (2003) Structure 11:825
Pan B, Xiong Y, Shi K, Deng J, Sundaralingam M (2003) Structure 11:815
Roitzsch M, Lippert B (2006) Angew Chem Int Ed 45:147
Roitzsch M, Lippert B (2004) Inorg Chem 43:5483
Freisinger E, Schimanski A, Lippert B (2001) J Biol Inorg Chem 6:378
Metzger S, Lippert B (1996) J Am Chem Soc 118:12467
Liu H, Gauld JW (2009) Phys Chem Chem Phys 11:278
Meyer M, Hocquet A, Sühnel J (2005) J Comput Chem 26:352
van Mourik T, Dingley AJ (2005) Chem Eur J 11:6064
Gu J, Leszczynski J (2002) J Phys Chem A 106:529
Sponer J, Spacková N (2007) Methods 43:278
Meyer M, Sühnel J (2008) J Phys Chem A 112:4336
Meyer M, Schneider C, Brandl M, Sühnel J (2001) J Phys Chem A 105:11560
Gu J, Leszczynski J (2001) Chem Phys Lett 335:465
Sponer J, Jurecka P, Hobza P (2004) J Am Chem Soc 126:10142
Jurecka P, Hobza P (2003) J Am Chem Soc 125:15608
Jurecka P, Sponer J, Cerny J, Hobza P (2006) Phys Chem Chem Phys 8:1985
van der Wijst T, Fonseca Guerra C, Swart M, Bickelhaupt FM (2006) Chem Phys Lett 426:415
Fonseca Guerra C, Bickelhaupt FM, Snijders JG, Baerends EJ (2000) J Am Chem Soc 122:4117
Zhao Y, Truhlar DG (2005) Phys Chem Chem Phys 7:2701
Swart M, van der Wijst T, Fonseca Guerra C, Bickelhaupt FM (2007) J Mol Model 13:1245
Baerends EJ, Autschbach J, Bérces A, Bickelhaupt FM, Bo C, Boerrigter PM, Cavallo L, Chong DP, Deng L, Dickson RM, Ellis DE, van Faassen M, Fan L, Fischer TH, Fonseca Guerra C, van Gisbergen SJA, Götz AW, Groeneveld JA, Gritsenko OV, Grüning M, Harris FE, van den Hoek P, Jacob CR, Jacobsen H, Jensen L, van Kessel G, Kootstra F, Krykunov MV, van Lenthe E, McCormack DA, Michalak A, Neugebauer J, Nicu VP, Osinga VP, Patchkovskii S, Philipsen PHT, Post D, Pye CC, Ravenek W, Rodriguez JI, Ros P, Schipper PRT, Schreckenbach G, Snijders JG, Solà M, Swart M, Swerhone D, te Velde G, Vernooijs P, Versluis L, Visscher L, Visser O, Wang F, Wesolowski TA, van Wezenbeek EM, Wiesenekker G, Wolff SK, Woo TK, Yakovlev AL, Ziegler T Theoretical Chemistry. ADF2008.01, SCM, Vrije Universiteit, Amsterdam, The Netherlands. http://www.scm.com
te Velde G, Bickelhaupt FM, Baerends EJ, Fonseca Guerra C, van Gisbergen SJA, Snijders JG, Ziegler T (2001) J Comput Chem 22:931
Fonseca Guerra C, Visser O, Snijders JG, te Velde G, Baerends EJ (1995) Fgtksdhfgsd. In: Clementi E, Corongiu G (eds) Methods and techniques for computational chemistry. STEF, Cagliari, pp 305–395
Baerends EJ, Ellis DE, Ros P (1973) Chem. Phys 2:41
Baerends EJ, Ros P (1975) Chem Phys 8:412
Baerends EJ, Ros P (1978) Int J Quantum Chem Symp 12:169
Fonseca Guerra C, Snijders JG, te Velde G, Baerends EJ (1998) Theor Chem Acc 99:391
Boerrigter PM, te Velde G, Baerends EJ (1988) Int J Quantum Chem 33:87
te Velde G, Baerends EJ (1992) J Comput Phys 99:84
Snijders JG, Vernooijs P, Baerends EJ (1981) At Nucl Data Tables 26:483
Krijn J, Baerends EJ (1984) Fit-functions in the HFS-method. Internal Report (in Dutch). Vrije Universiteit, Amsterdam
Versluis L, Ziegler T (1988) J Chem Phys 88:322
Swart M, Bickelhaupt FM (2006) Int J Quantum Chem 106:2536
Swart M, Bickelhaupt FM (2008) J Comput Chem 29:724
Grimme S (2004) J Comput Chem 25:1463
Grimme S (2006) J Comput Chem 27:1787
Antony J, Grimme S (2006) Phys Chem Chem Phys 8:5287
Elstner M, Hobza P, Frauenheim T, Suhai S, Kaxiras E (2001) J Chem Phys 114:5149
Wu X, Vargas MC, Nayak S, Lotrich V, Scoles G (2001) J Chem Phys 115:8748
Wu Q, Yang W (2002) J Chem Phys 116:515
Zimmerli U, Parrinello M, Koumoutsakos P (2004) J Chem Phys 120:2693
Becke AD (1988) Phys Rev A 38:3098
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785
Perdew JP (1986) Phys Rev B 33:8822 (Erratum: (1986) Phys Rev B 34:7406)
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865
Zhao Y, Truhlar DG (2008) Theor Chem Acc 120:215
Becke AD (1993) J Chem Phys 98:5648
Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ (1994) J Phys Chem 45:11623
Bérces A, Dickson RM, Fan L, Jacobsen H, Swerhone DP, Ziegler T (1997) Comput Phys Commun 100:247
Jacobsen H, Bérces A, Swerhone D, Ziegler T (1997) Comput Phys Commun 100:263
Wolff SK (2005) Int J Quantum Chem 104:645
Boys SF, Bernardi F (1970) Mol Phys 19:553
Klamt A, Schüürmann G (1993) J Chem Soc Perkin Trans 2:799
Klamt A (1995) J Phys Chem 99:2224
Pye CC, Ziegler T (1999) Theor Chem Acc 101:396
Swart M, Rösler E, Bickelhaupt FM (2007) Eur J Inorg Chem 3646
Nicu VP, Neugebauer J, Baerends EJ (2008) J Chem Phys A 112:6978
Jalkanen KJ, Degtyarenko IM, Nieminen, Cao X, Nafie LA, Zhu F, Barron (2008) Theor Chem Acc 119:191
Losada M, Xu Y (2007) Phys Chem Chem Phys 9:3127
Weise CF, Weisshaar JC (2003) J Phys Chem B 107:3265
Poon CD, Samulski ET, Weise CF, Weisshaar JC (2000) J Am Chem Soc 122:5642
Han WG, Jalkanen KJ, Elstner M, Suhai S (1998) J Phys Chem B 102:2587
Jalkanen KJ, Suhai S (1996) Chem Phys 208:81
Riley KE, Vondrasek J, Hobza P (2007) Phys Chem Chem Phys 9:5555
Kabelac M, Sherer EC, Cramer CJ, Hobza P (2007) Chem Eur J 13:2067
Kabelac M, Valdes H, Sherer EC, Cramer CJ, Hobza P (2007) Phys Chem Chem Phys 9:5000
Acknowledgments
We thank the International Max Planck Research School in Chemical Biology (IMPRS-CB) in Dortmund for a doctoral fellowship for T.v.d.W, the National Research Combination-Catalysis (NRSC-C) for a postdoctoral fellowship for C.F.G., the Spanish Ministerio de Educación y Ciencia (MEC) for a Ramon y Cajal fellowship for J.P., the Catalan Institution for Research and Advanced Studies (ICREA) for a research fellowship for M.S. and the Netherlands Organization for Scientific Research (NWO-CW and NWO-NCF) for further financial support. We thank one of the anonymous reviewers for the suggestion to use the M06-2X functional.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Dedicated to Professor Sandor Suhai on the occasion of his 65th birthday and published as part of the Suhai Festschrift Issue.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Fonseca Guerra, C., van der Wijst, T., Poater, J. et al. Adenine versus guanine quartets in aqueous solution: dispersion-corrected DFT study on the differences in π-stacking and hydrogen-bonding behavior. Theor Chem Acc 125, 245–252 (2010). https://doi.org/10.1007/s00214-009-0634-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00214-009-0634-9